
| Vogt–Koyanagi–Harada disease | |
|---|---|
 | |
| Especialidade | oftalmologia |
| Classificação e recursos externos | |
| CID-10 | H20.82, H30.81, H20.8, H30.8 |
| CID-9 | 364.24, 363.22 |
| CID-11 | 1827767178 |
| DiseasesDB | 13983 |
| eMedicine | 1229432 |
| MeSH | D014607 |
|
| |
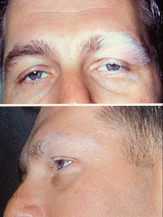
Síndrome de Vogt–Koyanagi–Harada doença (VKH), também conhecida como uveomeningoencefalite, é uma doença autoimune que afeta tecidos com melanina (melanócitos). O sintoma mais característico é a inflamação de uma das camadas de ambos olhos (uveíte bilateral difusa). VKH pode também afetar a orelha interna, reduzindo a audição, e afetar a pele e as meninges do sistema nervoso central.
Sinais e sintomas
Visão geral
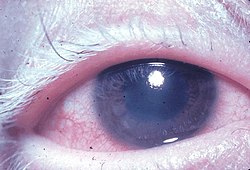
Essa doença é caracterizada por uveíte bilateral difusa, com dor, vermelhidão e embaçamento da visão. Os sintomas oculares podem ser acompanhadas por uma variável constelação de sintomas sistêmicos, tais como auditivas (zumbido, vertigem, e hipoacusia), neurológicas (meningismus, mal-estar, febre, dor de cabeça, náuseas, dor abdominal, rigidez do pescoço e nas costas, ou de uma combinação destes fatores. Sintomas menos frequentes incluem meningite, pleocitose em LCR, paralisia facial ou hemiparesia, mielite transversa e ciliar ganglionitis), e manifestações cutâneas, incluindo descoloração da pele similar a vitiligo e alopecia. O vitiligo é frequentemente encontradas na região do sacro.
Causa
Pode ser desencadeado por alguma infecção viral ou por trauma na pele ou dos olhos, sendo difícil determinar qual foi a causa da doença e prevenir. No entanto, VKH é atribuída uma reação autoimune: os próprios linfócitos T destruem antígenos encontrados nos melanócitos. Estimulado pela interleucina-23 (IL-23), Linfócitos T auxiliares e citocinas , como a interleucina-17 (IL-17) atacam algumas das proteínas nos melanócitos.
Fatores de risco
Os indivíduos afetados estão normalmente entre 20 e 50 anos de idade. Afeta duas vezes mais mulheres que homens. Normalmente não há nenhuma história de cirúrgia ou trauma ocular recente. VKH é mais comum entre asiáticos, latinos, árabes, índios americanos e mestiços. É muito menos comum em brancos e negros.
Tratamento
A uveíte aguda por VKH é geralmente sensível a altas doses orais de corticosteróides. Administração parenteral normalmente não é necessária. No entanto, complicações oculares podem exigir um subtenon ou intra -vítrea de injeção de corticosteroides ou bevacizumabe. No refratário situações, outros imunossupressores , como a ciclosporina, ou o tacrolimus, antimetabolitos (como azatioprina, micofenolato de mofetil ou metotrexato), ou agentes biológicos, tais como imunoglobulinas intravenosas (IVIG) ou infliximab podem ser úteis.
Prognóstico
O prognóstico geralmente é bom, com rápido diagnóstico e tratamento agressivo com imunomoduladores. Sintomas auditivos geralmente respondem à terapia com corticosteroides dentro de semanas a meses; a audição normalmente recupera-se completamente. Síntomas oculares mais crônicos como catarata, glaucoma ou atrofia óptica podem ocorrer. As alterações da pele geralmente persistem apesar do tratamento.
Epônimo
A Síndrome de VKH é uma homenagem aos oftalmologistas Alfred Vogt , da Suíça, e Yoshizo Koyanagi e Einosuke Harada do Japão. Vários autores, incluindo o árabe médico Mohammad al-Ghâfiqî no século XII, bem como Jacobi, Nettelship e a doença de Tay no século XIX, tinha descrito poliosis, neuralgias e distúrbios da audição. Esta constelação foi, provavelmente, muitas vezes devido a oftalmia simpática , mas provavelmente incluídos exemplos de VKH. Koyanagi fez a primeira descrição da doença em 1914, mas foi precedido por Jujiro Komoto, Professor de Oftalmologia da Universidade de Tóquio, em 1911. Apenas em 1929, associaram Koyanagi com a doença. Harada, em 1926, foi reconhecido por sua descrição abrangente do que agora conhecido como Vogt–Koyanagi–Harada doença.