
| Glicogenose | |
|---|---|
 | |
| Especialidade | endocrinologia |
| Classificação e recursos externos | |
| CID-10 | E74.0 |
| CID-9 | 271.0 |
| CID-11 | 1187107383 |
| MeSH | D006008 |
|
| |
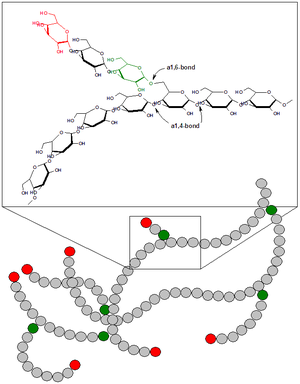
Glicogenose (também chamada de doença do armazenamento de glicogênio) é qualquer doença relacionada a erros inatos do metabolismo, resultantes de deficiências enzimáticas, que afetam o processamento da síntese do glicogênio ou sua quebra nos músculos e fígado.
Doenças de armazenamento do glicogênio resultam da incapacidade do indivíduo metabolizar o glicogênio decorrente de defeitos nas enzimas envolvidas no metabolismo do mesmo. É um nome genérico que engloba pelo menos diversas doenças hereditárias raras de armazenamento de glicogênio nos tecidos
Metabolismo normal do glicogênio: a síntese e degradação do glicogênio envolvem conjuntos separados de enzimas, funcionando de forma irreversível, ou seja, o processo de degradação não é o inverso da síntese. Pelo menos 8 enzimas estão envolvidas neste processo. Basta a ausência de uma para que a síntese ou a degradação fique comprometida, ou a molécula de glicogênio pode ser anormal. Além disso, há enzimas que têm formas distintas em órgãos diferentes.
Na década de 70 os pacientes com glicogenose apresentavam um mortalidade elevada e danos neurológicos permanentes com grande atraso no desenvolvimento mental e crescimento. O tratamento atual tem alterado significativamente o curso clínico, e houve notável melhora do prognóstico nos pacientes com glicogenose tipo I, com expectativa de vida ultrapassando a terceira década.
Tipos de Glicogenoses
Existem doenças que geralmente são consideradas como glicogenoses, conforme o defeito enzimático específico e os órgãos afetados. Cada tipo de glicogenose é causado por diferentes genes e diferentes enzimas, que acarretam em variações nas manifestações clínicas e nas formas de tratar as doenças.
| Tipos | Enzima Deficiente | Gene |
Características
Clínicas |
Tratamento | Nome da Doença |
| Glicogenose Tipo 0 | Glicogênio sintetase (hepática) | GYS2 | Hipoglicemia cetótica ao jejum;
Hiperglicemia; Hiperlactacidemia; Hiperlipidemia pós-prandial; e Ausência de hepatomegalia. |
Dieta hiperproteica e com carboidratos complexos de baixo índice glicêmico;
Uso noturno de amido de milho cru. |
--- |
| Glicogenose Tipo Ia | Glicose-6-fosfatase | G6PC | Hipoglicemia;
Hepatomegalia; Retardo do crescimento; Acidose lática; Hiperuricemia; e Hiperlipidemia. |
Uso de amido de milho cru;
Restrição de galactose, frutose, lactose e sacarose. |
Doença de Von Gierke |
| Glicogenose Tipo Ib | Transportador de Glicose-6-fosfato | SLC37A4 | Hipoglicemia;
Hepatomegalia; Retardo do crescimento; Acidose lática; Hiperuricemia; Hiperlipidemia. Acompanhado de neutropenia (diminuição do número de neutrófilos no hemograma); Infecções bacterianas recorrentes; e Pode ocorrer doença inflamatória intestinal. |
Uso de amido de milho cru;
Restrição de galactose, frutose, lactose e sacarose. Uso de estimulador de colônia de neutrófilos e tratamento da doença inflamatória intestinal. |
--- |
| Glicogenose Tipo II | Alfa-glicosidase | GAA | Pode ter apresentação infantil, com hipotonia, dificuldade de sucção e cardiomiopatia; ou
Apresentação adulta com doença muscular progressiva, cardiomiopatia e dificuldades respiratórias. |
Fisioterapia motora e respiratória; e
Terapia de reposição enzimática. |
Doença de Pompe |
| Glicogenose Tipo IIIa e IIIb | Enzima desramificadora de glicogênio | AGL | Hepatomegalia;
Hipoglicemia cetótica; Retardo do crescimento; Hiperlipidemia; Elevação da AST e ALT, CPK. Fraqueza muscular e cardiomiopatia ocorrem no subtipo III a. |
Uso de amido de milho cru;
Dieta hiperproteica; e Restrição de sacarose. |
Doença de Cori ou Doença de Forbe |
| Glicogenose Tipo IV |
amilo-1,4-1,6-glicosidase
Enzima ramificadora de glicogênio |
GBE1 | Hepatomegalia;
Retardo do crescimento; e Cirrose. |
Transplante de fígado nos casos graves. | Doença de Andersen |
| Glicogenose Tipo V | glicogênio fosforilase muscular | --- | Intolerância ao exercício, mialgias e crises de mioglobinúria por rabdomiólise. | Exercício físico controlado. | Doença de McArdle |
| Glicogenose Tipo VI | glicogênio fosforilase hepática | PYGL | Hepatomegalia;
Retardo do crescimento; Hipoglicemia; Hiperlipidemia e hipercetose leves. |
Se sintomático, aumento de carboidrato e alimentação frequente e dieta hiperproteica. | Doença de Hers |
| Glicogenose Tipo VII |
fosfofrutoquinase muscular
Phosfofrutoquinase-1 |
PFKM | Miopatia, contraturas articulares, epilepsia, atraso de desenvolvimento, catarata. Na forma adulta, pode se apresentar com fraqueza muscular progressiva. | Dieta rica em proteínas e vitamina B6; evitar exercício extenuante. | Doença de Tarui |
| Glicogenose Tipo IXa | Fosforilase quinase (subunidade alfa) | PHKA2 | Hepatomegalia;
Hipoglicemia cetótica ao jejum; Retardo do crescimento; Elevação de AST/ALT, hiperlipidemia leves e cetonas aumentadas no sangue e urina. Podem haver sintomas musculares e elevação da enzima muscular CpK. |
Uso de amido de milho cru; dieta rica em proteínas; evitar grandes quantidades de sacarose. | --- |
| Glicogenose Tipo IXb | Fosforilase quinase (subunidade beta) | PHKB | Hepatomegalia, hipoglicemia cetótica ao jejum, retardo do crescimento, elevação de AST/ALT e hiperlipidemia leves. | Uso de amido de milho cru; dieta rica em proteínas; evitar grandes quantidades de sacarose. | --- |
| Glicogenose Tipo IXc | Fosforilase quinase (subunidade gama) | PHKG2 | Hepatomegalia, hipoglicemia cetótica ao jejum, retardo do crescimento, elevação de AST/ALT e hiperlipidemia leves. Cirrose hepática pode se desenvolver. | Uso de amido de milho cru; dieta rica em proteínas; evitar grandes quantidades de sacarose. | --- |
| Glicogenose Tipo X | Fosfoglicerato mutase | --- | Intolerância aos exercícios, cãibras, mioglobinúria e fraqueza | --- | --- |
| Glicogenose Tipo XI |
transportador de glicose
Transportador de glicose-2 |
GLUT2 | Hipoglicemia, déficit de crescimento, raquitismo e abdome protuberante devido ao aumento do tamanho do fígado e rins. | Restrição da ingestão de galactose, suplementação de água, amido de milho, eletrólitos e vitamina D. | Doença de Fanconi-Bickel |
| Glicogenose Tipo XII | Aldolase A | --- | Intolerância ao exercício e fraqueza após doença febril.
Fraqueza muscular precoce e febre; Episódios de rabdomiólise com elevações da creatina quinase, aspartato aminotransferase, alanina aminotransferase, lactato desidrogenase e bilirrubina; diminuição na concentração de hemoglobina e hematócrito (relatos) |
--- | --- |
| Glicogenose Tipo XIII | Beta-enolase (ENO3) | --- | Intolerância ao exercício, cãibras. | --- | --- |
| Glicogenose Tipo XV | Glycogenin-1 (GYG1) | --- | Atrofia muscular | --- | --- |
HERANÇA GENÉTICA
Todas as glicogenoses hepáticas (exceção da IX alfa) possuem um padrão de herança autossômico recessivo, com risco de recorrência de 25% para as próximas gerações, independentemente do sexo da criança ou de já ter outros filhos que tenham a doença. A transferência do gene para os filhos independe também dos pais serem parentes.
A chance de uma pessoa com glicogenose ter um filho/filha também com glicogenose vai depender se o outro genitor possui ou não o gene da glicogenose. Se o outro genitor também possuir glicogenose, a recorrência é de 100% do filho/filha ter glicogenose. Se o outro genitor não possuir nenhum gene afetado, todos os filhos receberão um gene afetado e um normal, sendo portadores de um gene, mas sem desenvolver a doença. Mas se o outro genitor possuir um gene afetado e um normal, a chance é de 50% receberem um gene normal e um afetado, sendo portadores somente, e 50% de receber os dois genes afetados, possuindo glicogenose.
A exceção ocorre com a glicogenose tipo IX, que possui um padrão de herança ligado ao cromossomo X, em que a mãe portadora passa o cromossomo X afetado para o filho. Nesse caso, se for menina, ela tem 50% de chance de não receber o gene afetado e ser normal e 50% de chance de receber o X afetado da mãe e ser também portadora. Já se for menino, ele tem 50% de chance de ser normal, e 50% de chance de receber o X afetado da mãe, e possuir glicogenose.
DIAGNÓSTICO
Variam de acordo com o tipo de glicogenose. O diagnóstico é clínico, baseado nos sintomas, contudo para confirmação do diagnóstico e definição do tipo de Glicogenose, relevante para o planejamento da terapia, torna-se importante a realização do teste genético.
Cada tipo de glicogenose possui características e tratamentos diferentes. É de extrema importância conhecer o tipo específico para não realizar o tratamento de forma errada e agravar o quadro de saúde.
Exames
Glicose;
Lactato;
Ácido Úrico;
Triglicérides;
Colesterol; e
Ultrassom do Fígado.
O teste do Glucagon não é indicado e pode ser perigoso, pois não elevará o nível de glicose e vai elevar significativamente o ácido lático, podendo provocar hipoglicemias graves e crise convulsiva.
EPIDEMIOLOGIA
No geral, de acordo com um estudo na Colúmbia Britânica, aproximadamente 2,3 crianças a cada 100.000 nascimentos têm alguma forma de doença de armazenamento de glicogênio. Nos Estados Unidos, estima-se que ocorram em 1 a cada 20.000 - 25.000 nascimentos. A taxa de incidência holandesa é estimada em 1 em cada 40.000 nascimentos.
As glicogenoses são consideradas doenças raras, considerando a frequência estimada global de 1/20.000 a 1/25.000 nascidos vivos.
Os tipos mais comuns são os I, II, III e IX. No Brasil não há um levantamento preciso da incidência de cada tipo, mas acredita-se que os tipos mais comuns sejam os tipos I e III (dados obtidos de levantamento dos pacientes do Hospital de Clínicas de Porto Alegre e da Universidade de Campinas).
Ver também
- Genética
- Hipoglicemia
- Síndrome de Fanconi
- Erro metabólico hereditário
- Doenças associadas à deficiência mental
- ABGLICO. ASSOCIAÇÃO BRASILEIRA DE GLICOGENOSE: o que é glicogenose. O que é Glicogenose. 2019. Disponível em: https://abglico.com.br/. Acesso em: 02 maio 2021.
- Kishnani, P.S., Goldstein, J., Austin, S.L. et al. Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 21, 772–789 (2019). https://doi.org/10.1038/s41436-018-0364-2.
- CENTER, University Of Pittsburgh Medical. DEPARTMENT OF PATHOLOGY: case 751 -- a 3 year old male with fatigue and hepatomegaly. Case 751 -- A 3 year old male with fatigue and hepatomegaly. 2020. Disponível em: https://path.upmc.edu/. Acesso em: 02 maio 2021.