
| Parte de uma série sobre |
| Evolução |
|---|
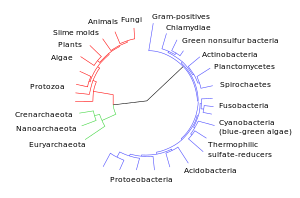 Diagrama da divergência dos grupos taxonómicos modernos em relação aos seus ancestrais comuns.
|
|
Tópicos fundamentais
|
Evolução molecular refere-se a vários processos evolutivos resultantes de alterações no DNA, RNA, e/ou Proteína. A evolução no ácido nucleico ocorre por processos mutacionais, os quais, por sua vez, provocam alterações nos aminoácidos codificados. Aminoácidos alterados resultam numa subsequente alteração na forma e funcionamento das proteínas. A evolução também pode ocorrer no nível do genoma, por processos como a duplicação de genes e transferência horizontal de genes. Quando um destes processos resultar em mudanças significativas na história das espécies, ocorrerá um processo evolutivo que gerará diferenças entre a espécie e seu ancestral, fato que representa a base para a evolução dos organismos. Todas essas informações sobre genes e proteínas podem ser utilizadas pela filogenética molecular para estabelecer uma árvore Filogenética concernente à evolução do grupo em estudo.
A área emergiu como um campo científico nos anos 60, quando pesquisadores das áreas da biologia molecular, biologia evolutiva e genética de populações procuraram entender algumas descobertas que haviam sido feitas sobre a estrutura e função de ácidos nucléicos e proteínas. Como disciplina, o campo da Evolução Molecular se encarrega da evolução de genes e proteínas, dando ênfase à taxa de mutação e aos mecanismos que regem a evolução molecular. Uma das teorias neste campo é a teoria neutralista da evolução, desenvolvida por Motoo Kimura.
A Evolução Molecular permite revelar a natureza das mudanças no material genético e nos produtos codificados por ele, além de determinar por métodos estatísticos o curso histórico-evolutivo dessas mudanças em uma determinada linhagem, ou entre linhagens diferentes.
Causas da evolução molecular
Mutações
Mutação é um erro que ocorre no material genético, principalmente durante o processo de replicação do DNA, envolvendo falhas nos mecanismos normais pelos quais os genomas são copiados e, eventualmente, corrigidos. Além disso, a mutação pode também ocorrer por meio de elementos transponíveis (transposão), e por meio de outros fatores, como radiação ultravioleta, e agentes mutagênicos. As mutações podem acontecer tanto células somáticas quanto em germinativas. Ao contrário do que acontece em células germinativas, as mutações que ocorrem em células somáticas não são hereditariamente transmissíveis e não influenciam no processo evolutivo.
A classificação de uma mutação depende do aspecto que é utilizado para a análise. Os dois tipos principais para o estudo evolutivo têm em conta o tipo de alteração estrutural que produzem - mutação pontual, inserção (genética), deleção -, e aquelas classificadas pela perda (não-sinônimas) ou não (sinônimas) de função do gene ou aminoácido alterado. Uma classificação não exclui a outra, ou seja, uma mutação caracterizada como pontual pode gerar um resultado que possa a classificar como sinônima, por exemplo. As classificações são apenas maneiras diferentes de se referir a uma determinada alteração no material genético.
A resposta adaptativa e evolutiva à mutação dependerá das consequências funcionais resultantes dessa alteração. A seleção natural irá agir a partir do momento em que a mutação resultar em alguma alteração prejudicial ou vantajosa para o organismo: se o efeito prejudicial for importante, a alteração será eliminada, podendo, em casos mais extremos, resultar na morte do organismo portador, sem deixar descendentes. Ao contrário, se a mutação conferir vantagem para o organismo, ela será selecionada positivamente, podendo ser acumuladas ao longo das gerações, conferindo vantagens adaptativas para os portadores. As mutações neutras, de acordo com Kimura, seriam aquelas alterações que não provocariam o aparecimento de características adaptativas, ou seja, nem prejudiciais e nem benéficas. Porém, esse tipo de mutação também é distribuída e pode se fixar na população, mas nesse caso será por deriva genética. Situação parecida acontece com as mutações sinônimas e não-sinônimas: no primeiro caso, as mutações irão interferir pouco ou nada no processo de adaptação do indivíduo, já que apresentam um impacto funcional não significativo. Dessa forma, tendem a evoluir muito mais rápido por escaparem dos efeitos da seleção natural negativa, que elimina mutações deletérias que interferem com a função dos genes e proteínas. Um exemplo de mutações sinônimas são aquelas que ocorrem na terceira posição do códon, que, devido ao código genético ser degenerado, não resultarão em alteração do aminoácido correspondente ao novo códon. Já no segundo caso, ocorrerá forte ação da seleção natural, pelo fato das mutações gerarem alterações favoráveis ou desfavoráveis no genoma. Essa última situação pode ser exemplificada pelas mutações que acontecem na primeira ou terceira posição do códon e que acabam alterando o aminoácido correspondente ao novo códon.
Recombinação
A recombinação genética nos eucariotas envolve a troca de material genético entre cromossomos homólogos; já nos procariotas e vírus, a recombinação envolve a adição de fragmentos do respectivo DNA em outro genoma. Ou seja, esse processo representa o processo de reorganização dos genes já existentes: a mutação cria a novidade genética em um indivíduo e a recombinação distribui para os seus descendentes. Devido à existência dessa reorganização gênica, o processo de recombinação é capaz de produzir, em organismos sexuados, grande variação genética entre os descendentes e entre os descendentes e seu ancestral. Em uma população, a variação produzida pela recombinação genética é fundamental para promover a evolução dos organismos em resposta a alterações ambientais.
Duplicação Gênica
O aumento na quantidade de cópias de um determinado fragmento de DNA pode ser feito por meio da duplicação gênica, caracterizado como um processo que pode envolver parte de um gene, partes de um cromossomo ou um genoma inteiro, os quais poderão ser duplicados por crossing over desigual, retrotransposição, aneuploidia e poliploidia. Uma vez duplicado, um gene pode estar sujeito a três destinos: O primeiro consiste na situação em que uma das duas cópias do gene degenera em um pseudogene, o qual pode ser subsequentemente excluído do genoma. O segundo ocorre quando o gene duplicado mantém a sua função original, possibilitando ao organismo produzir uma maior quantidade de RNAs. O terceiro ocorre quando a cópia duplicada assume uma nova função; nesse caso, o gene duplicado acumula mudanças moleculares ao longo do tempo, resultando em uma nova função, e o original mantém a sua função (ou uma função bem similar à original, já que ele também pode ser alterado por mudanças evolutivas). Determinando qual dessas possibilidades de fato ocorreu com o determinado gene em estudo, é possível verificar se esse gene apresenta algum outro gene homólogo (os genes são homólogos entre si quando eles são derivados de um ancestral comum), e obter mais informações evolutivas à respeito das suas linhagens.
Transferência Horizontal de Genes
A transferência horizontal de genes (THG) ocorre quando um gene de um genoma de uma espécie é copiado para o genoma de outra espécie. Estas transferências ocorrem principalmente entre procariotas, constituindo uma importante ferramenta na sua adaptação ao um nicho específico, pois a aquisição de um genoma melhorado aumenta a adaptabilidade destes organismos. Contudo, o impacto da THG nos seres procariotos irá depender principalmente do número de genes que foram transferidos e se foram mantidos no genoma com sucesso. O problema ocorre durante os estudos filogenéticos que envolvem apenas um gene. Nesse caso, organismos de taxa diferentes que tenham sido envolvidos em uma transferência horizontal de genes podem ser erroneamente considerados com grupos irmãos. Por outro lado, a THG entre células eucarióticas entre diferentes espécies ainda não é muito estudada, mas acredita-se que seja um evento raro, não parecendo ter apresentado um papel significante na evolução eucariótica.
Origem e evolução das macromoléculas
As macromoléculas são representadas pelo RNA, DNA e pelas proteínas, e os processos evolutivos que levaram ao seu surgimento devem ter ocorrido antes da evolução da célula, já que esta, mesmo sendo a unidade básica dos organismos, não poderia ter evoluído sem antes terem sido originadas as moléculas que constituiriam as suas estruturas. De acordo com a teoria da Origem da Vida, uma evolução química teria feito com que compostos orgânicos correspondentes aos aminoácidos, nucleotídeos e açúcares, fossem formados pela síntese abiótica, nas condições climáticas da Terra primitiva. A formação de compostos maiores seria resultado da condensação daquelas primeiras moléculas: nucleotídeos se ligaram por meio de ligações fosfodiéster, resultando na formação de ácidos nucléicos, enquanto que ligações amida uniram os aminoácidos, formando proteínas.
Coloca-se a questão de ter sido o DNA ou o RNA quem surgiu primeiro durante a evolução das macromoléculas. Num primeiro momento, pode-se concluir que, como o DNA representa o grupo de moléculas que detém as informações genéticas, então ele é a molécula que deveria ter surgido primeiro, para então originar o RNA e as proteínas. Porém, pesquisas indicam exatamente o contrário. Elas afirmam que os RNA foram as primeiras moléculas a aparecerem. Chegou-se a essa conclusão a partir de várias evidências: (a) A de que RNA apresenta uma tripla função: a de mensageiro, de transportador e ribossômico; isso mostra que ele teria capacidade de manter certo nível de organização celular.
(b) Descoberta de RNA com atividade catalítica, podendo catalisar reações químicas na célula primitiva. Essa propriedade era considerada exclusiva das proteínas. No entanto, a melhor capacidade catalisadora das proteínas indica que estas não poderiam ter surgido antes do RNA, já que este apresenta uma atividade catalítica inferior.
(c) Presença de algumas moléculas idênticas ou muito semelhantes aos monômeros de RNA em todos os seres vivos. Isso indica que o RNA já estava presente no ancestral comum dos seres vivos.
(d) O fato dos desoxirribonucleotídeos serem derivados dos ribonucleotídeos, ou seja, a unidade principal formadora do DNA seria derivada da unidade formadora do RNA.
(e) Capacidade de armazenamento de informação genética, o que prova que o DNA não precisaria estar presente para que o organismo fosse capaz de armazenar material genético.
(f) Ribonucleotídeos apresentam papel fundamental no metabolismo, principalmente na forma de ATP, indicando que os principais aspectos do metabolismo já estavam estabelecidos antes da evolução do DNA.
Apesar de todas essas características levarem a crer que o RNA é suficiente para formar um grupo de catalisadores autorreplicantes, provavelmente o RNA não terá sido a primeira molécula a fazer isso. Quimicamente, é improvável que longas moléculas de RNA possam ter sido formadas inicialmente por sistemas completamente não-enzimáticos, visto que: os ribonucleotídeos são difíceis de serem formados não-enzimaticamente; e o fato de que, para a formação do RNA, são necessárias várias ligações fosfodiéster 3’-5’ sejam formadas ao invés de outras ligações, como hidrólises, ligações 2’-5’ e 5’-5’. Dessa forma, a situação mais provável é de que as primeiras moléculas a possuírem atividade catalítica e de armazenamento de informações tenham sido polímeros semelhantes ao RNA, mas quimicamente mais simples.
Provavelmente a molécula de DNA surgiu posteriormente ao evento de surgimento da síntese protéica, já que proteínas podem ser sintetizadas na ausência de DNA, mas não de RNA. Além disso, precedendo o surgimento dessa molécula, também deve ter ocorrido o surgimento do processo de Transcriptase reversa, e do desenvolvimento de processos enzimáticos capazes de converter precursores de ribonucleotídeos em precursores de desoxirribonucleotídeos. Depois do seu surgimento, o DNA foi favoravelmente selecionado devido a várias razões. Dentre elas destacam-se: A molécula dupla fita é extremamente resistente e estável. Sendo mais estáveis, o DNA pode aumentar de tamanho através do processo de duplicação gênica. Apresenta a informação duplicada, em cada uma das fitas de DNA, o que também facilita o reparo em caso de dano de uma das fitas, já que a fita complementar apresentará a mesma informação.
Evolução na estrutura proteica
Um aminoácido é codificado a partir de um códon específico, que por sua vez é formado por uma combinação específica de três nucleotídeos. Qualquer alteração (principalmente por mutação pontual) desses nucleotídeos pode resultar na alteração do aminoácido a ser produzido. Porém, a existência de somente 20 aminoácidos permite que o código genético seja degenerado, ou seja, um aminoácido pode ser produzido a partir de vários códons específicos a ele. Normalmente, alteração na terceira posição do códon não causa alteração no aminoácido a ser traduzido, ao contrário do que acontece quando ocorre alteração nas outras posições.
Proteínas são formadas por domínios, que são fragmentos com estrutura, função e/ou história evolutiva diferente. Esses domínios podem ocorrer sozinhos, formando proteínas de um único domínio, mas na maioria das vezes as proteínas serão formadas pela combinação entre vários desses fragmentos, resultando em uma grande cadeia polipeptídica. Durante a evolução, proteínas foram modificadas por meio da fusão e recombinação de domínios, além da diferenciação daqueles já existentes.
A recombinação desses domínios pode ser resultado da recombinação entre os éxons correspondentes, evento que ocorre durante o embaralhamento de éxons. Nesse processo, os íntrons, por corresponderem a maior parte do genoma, representam os sítios preferenciais onde os pontos de recombinação entre os éxons irão ocorrer.
Evolução da estrutura gênica

O gene eucariota atual tem a estrutura (Fig.1) composta por éxons (exão) e íntrons (iintrão), enquanto Archaea e Bacteria não apresentam íntrons. Uma das hipóteses é a de que os íntrons já existiam nos primeiros genes e foram posteriormente perdidos em Archaea e Bacteria. Em outras palavras: os genes são descendentes de minigenes, sendo que cada um representaria um éxon; enquanto que os íntrons descendem dos espaços entre esses minigenes e apresentariam capacidade de auto-splicing. Em Eucarya, eles teriam evoluído para íntrons spliceossomos.
A perda de íntrons pelos Archaea e Bacteria seria conseqüência de uma pressão evolutiva para a redução do tamanho do seu genoma, levando à diminuição do tempo de replicação. Isso representaria uma vantagem competitiva, já que menor tamanho do genoma representa menos tempo necessário para a reprodução e crescimento do organismo. Porém, essa hipótese representa um cenário não parcimonioso ao postular que uma característica genômica, que está ausente em dois dos três domínios da vida, seria ancestral a todos esses domínios.
A hipótese mais aceita é de que os genes antigos não apresentariam íntrons, e que a sua adição ao genoma teria ocorrido após o surgimento da célula eucariótica, ou após o processo de endossimbiose que deu origem às mitocôndrias. Os spliceossomos nucleares seriam derivados dos íntrons grupo II auto-splicing, que é o grupo presente nas mitocôndrias; então deve ter havido um ganho de íntrons em genes transferidos de organelas para o núcleo.
Além de alterações estruturais, ao longo do processo evolutivo o genoma também sofreu alteração de tamanho, havendo tanto eventos de redução quanto eventos de aumento do genoma. A diminuição do genoma ocorreu principalmente por processos de transferência ou perda de genes, e esses eventos foram bastante representativos no processo de endossimbiose que originou mitocôndrias/cloroplastos, e no processo evolutivo do parasitismo.
- Redução durante a endossimbiose – Vários genes das organelas eram provavelmente redundantes e consequentemente foram deletados, sem serem posteriormente substituídos; enquanto que outros foram transferidos para o genoma nuclear da célula, até então hospedeira.
- Redução no parasitismo – Durante o processo de parasitismo, o hospedeiro fornece ao parasita várias das suas necessidades metabólicas e fisiológicas. Dessa forma, durante o evento de coevolução desses dois organismos, o parasita sofreu redução do material genético, já que vários genes, relacionados a atividades metabólicas e fisiológicas, que estavam presentes nele, foram perdidos por não serem mais necessários.
Por outro lado, o aumento do genoma pode ocorrer por dois processos principais:
- Aumento global, no qual todo o genoma, ou grande parte dele, é duplicado. Ocorre principalmente por eventos de poliploidia;
- Aumento regional, na qual uma sequência especifica ou é duplicada, gerando regiões repetitivas; ou por transferência horizontal.
Adaptação Molecular
Existem três formas principais pelas quais uma mudança, a nível molecular, pode gerar adaptação: mutação, seleção natural e deriva genética.
- Mutação é a causa principal para a variação genética, mas consiste em uma força evolutiva fraca, não podendo, sozinha, alterar a frequência do mutante dentro da população. A maneira mais simples para alterar a frequência da mutação seria se ela surgisse independentemente e várias vezes, o que consiste um processo lento e improvável;
- Contudo, uma nova mutação pode sofrer pressão de forças evolutivas que façam com que aumente de frequência, resultando na sua fixação, ou diminua de frequência a ponto de ser eliminada da população. Se ela sofrerá aumento ou diminuição dependerá de dois fatores. O primeiro fator representa a ação que a mutação tem sobre os indivíduos, ou seja, se ela favorece ou desfavorece a sobrevivência e reprodução. Mutações vantajosas são aquelas que aumentam a adaptabilidade do organismo, enquanto que mutações deletérias diminuem a adaptabilidade, e as neutras não apresentam nenhum efeito adaptativo. O segundo fator que influencia é o tamanho da população em questão;
- O papel da Seleção Natural é aumentar a frequência das mutações benéficas até que elas sejam fixadas na população (seleção positiva), ou diminuir a frequência das deletérias para que elas sejam eliminadas (seleção negativa). A seleção não atuará nas mutações neutras;
- A deriva promoverá a variação de frequência das mutações aleatoriamente. Dessa forma, uma nova mutação precisará de muito mais tempo para se fixar quando a deriva, ao invés da seleção, atuar sobre ela.
Filogenética Molecular
A filogenética molecular é a área que estuda as relações evolutivas entre os organismos, utilizando dados moleculares, como sequências de DNA e de proteínas. Como vantagens em utilizar dados moleculares ao invés de morfológicos temos:
(a) DNA e Proteínas são entidades estritamente hereditárias. Isso pode não ser verdade para caracteres morfológicos, que muitas vezes podem ser influenciados por fatores ambientais.
(b) A descrição dos estados de caracteres moleculares é mais precisa.
(c) Evoluem de uma maneira mais regular, o que fornece informações mais claras sobre as relações evolutivas entre organismos.
(d) Dados moleculares podem ser discretos e assim servir melhor à análise estatística do que os dados morfológicos.
(e) Dados moleculares podem ser utilizados para estudar as relações evolutivas entre organismos distantemente relacionados.
(f) Sequências moleculares proporcionam uma grande quantidade de evidência, pois cada alteração de aminoácido ou de base pode ser considerada como uma evidência distinta.
Reconstrução de Filogenias
Escolha dos dados moleculares
O ideal é que a análise evolutiva de um grupo seja feita tanto por meio de sequências de DNA quanto de aminoácido. Porém, isso nem sempre será possível. Nesse caso, a decisão em utilizar ou DNA ou proteínas para a análise deve ser feita levando em consideração a velocidade de evolução de cada uma dessas moléculas e o tempo de divergência entre as linhagens que serão estudadas. Normalmente, as sequências de aminoácidos apresentam uma taxa de evolução mais lenta do que a dos nucleotídeos, então poderão ser utilizadas quando as sequências de nucleotídeos apresentarem alguma saturação. Portanto, para táxons distantemente relacionados, as sequências de DNA provavelmente irão ter perdido grande parte das informações filogenéticas. A melhor alternativa seria então a utilização e sequências de aminoácidos. A saturação ocorre quando várias mudanças ocorrem em um mesmo ponto, fazendo com que a informação filogenética dessa mudança seja perdida.
Durante o estudo dos dados escolhidos, surge outro problema: nem todos os caracteres para os quais existem evidências indicarão a mesma filogenia. Dessa forma, quando vários caracteres são utilizados para inferir a filogenia de um grupo, e nem todos resultarem em árvores iguais ou com grande nível de similaridade, provavelmente existe alguma informação errada com pelo menos um deles. Então a primeira coisa a se fazer depois de escolher os dados a serem analisados e comparados, é determinar quais dos caracteres fornecerão informações confiáveis e quais serão inconfiáveis; estes, por sua vez, deverão ser descartados da análise. Essa análise inicial deverá ser feita em três etapas principais:
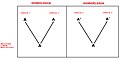

- Distinção entre homologias e homoplasias - Fig.2 - Uma homologia é um caráter compartilhado por várias linhagens e que estava presente no ancestral mais comum a elas, sendo, portanto, capaz de revelar relações filogenéticas entre os grupos estudados. Dessa forma, homologia é um caráter confiável. Homoplasia e um caráter compartilhado por várias linhagens, mas que não estava presente no ancestral mais comum a elas, e por isso não será capaz de revelar relações filogenéticas entre os grupos em questão. Homoplasia não é um caráter confiável porque ela pode ter surgido a partir de convergência evolutiva, ou seja, as linhagens estudadas podem ter evoluído características semelhantes porque sofreram pressões seletivas semelhantes, e, nesse caso, não existe relação de parentesco entre elas.
- Distinção entre homologias derivadas e ancestrais - Fig. 3 - Homologias ancestrais são características que estavam presentes no ancestral comum a todos os grupos estudados. As derivadas são características que evoluíram dentro do grupo em questão, ou seja, evoluíram após o ancestral comum. Somente as derivadas constituem evidências de que as duas espécies apresentam um ancestral comum mais recente, pois as homologias ancestrais serão compartilhadas também com outros grupos, já que terá surgido antes do ancestral comum mais recente ao grupo em estudo. A distinção entre as derivadas e as ancestrais implica que a árvore esteja polarizada e essa polarização pode ser feita de duas maneiras principais: I- Comparação com grupo externo: A polaridade do caráter, ou seja, a determinação de qual caráter é derivado e qual é o ancestral, é obtida por meio da observação de uma linhagem estreitamente relacionada, mas que ao mesmo tempo não pertence, filogeneticamente, ao grupo em estudo. Essa linhagem é chamada de grupo externo. Pela parcimônia, a condição do caráter que é encontrado no grupo externo e a provável condição ancestral no grupo em consideração; II- Comparação com registro fóssil: Pode-se inferir que eram as condições de caráter ancestral aquelas que são encontrados nos fósseis mais antigos.
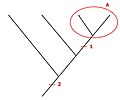
- Distinção entre homologia por duplicação ou por especiação – Fig. 4 - A partir de dois genes homólogos, tem-se que: os dois genes serão parálogos se eles foram originados a partir de um evento de duplicação; serão ortólogos quando foram originados como resultado de um processo de especiação. A utilização de um ou de outro dependerá da análise a ser feita. Cópias parálogas devem ser utilizadas para o estudo de eventos de duplicação gênica em famílias ou superfamílias de genes; cópias ortólogas devem ser utilizadas para a reconstrução filogenética de grupos taxonômicos.
Alinhamento de sequências
O alinhamento tem como objetivos fazer com que a posição de cada base (ou aminoácido) entre as diferentes sequências comparadas seja considerada homóloga. Isso é necessário para que se tenha certeza de que cada sítio de uma sequência corresponda ao mesmo sítio da outra. A partir daí será possível determinar as mudanças que ocorreram em todas as sequências.
Métodos de reconstrução na filogenia molecular
- Máxima parcimônia - O mais simples é o mais provável. A partir de um grupo de sequências alinhadas, várias são as árvores filogenéticas não polarizadas que podem ser construídas, e que retratarão a evolução do grupo. Porém, a árvore que provavelmente retratará mais fielmente o evento evolutivo será aquela com menor número de passos evolutivos, ou seja, aquela na qual foi necessário um conjunto menor de mudanças para chegar aos estados de caracteres encontrados no grupo estudado. Passos evolutivos, em análises moleculares, podem ser representados por substituições de bases na molécula de DNA, ou alterações nos aminoácidos.
Durante a análise dos dados, é importante detectar e eliminar aqueles caracteres que não são informativos. Um caráter não informativo é aquele que, para todas as árvores possíveis, irá requerer um mesmo número de passos evolutivos para chegar ao estado atual; então, nesse caso, não ocorre diferenciação significativa entre as árvores.
- Máxima verossimilhança - A partir de um modelo de evolução de sequências previamente determinado, o método da verossimilhança calcula a probabilidade de um conjunto de dados estar representando um processo que realmente ocorreu. Ou seja, é um método estatístico computacional que calcula a probabilidade de uma determinada árvore estar retratando a real filogenia do grupo. A filogenia será inferida achando-se a árvore com maior verossimilhança.
- Distâncias moleculares - Infere-se que as espécies com menores distâncias entre si estão mais estritamente relacionadas, então são agrupadas, filogeneticamente, em um mesmo grupo. Uma das maneiras de calcular a distância molecular entre linhagens é determinar o número de bases nucleotídicas que diferem entre elas; as duas linhagens que apresentarem um menor número de bases diferentes serão agrupadas juntas em relação às demais linhagens. Um problema desse método é que nem sempre muitas diferenças representam maior tempo desde a divergência, pois uma linhagem pode evoluir mais rápido do que a outra, e um mesmo sítio pode ter sofrido múltiplas alterações.
Independentemente do método utilizado, após selecionar a árvore que se supõe representar mais fielmente a evolução do grupo, é possível realizar testes de confiabilidade. A confiança é a probabilidade de que os membros de um dado clado sejam sempre membros desse clado. Um dos recursos que pode ser empregado é estabelecer o valor de bootstrap, que tem caráter estatístico é determina o suporte de cada nó das árvores filogenéticas, ou seja, a probabilidade daquele nó realmente retratar a monofilia do grupo.
Relógio Molecular
A Teoria do Relógio Molecular estabelece que o DNA e as proteínas evoluem em taxas relativamente constantes entre diferentes organismos. Devido a essa constância, infere-se que diferenças genéticas entre duas espécies são proporcionais ao tempo decorrido desde que elas divergiram a partir do ancestral comum mais recente. Ao utilizar o relógio molecular em análises filogenéticas, é importante a escolha de sequências que apresentem a mesma taxa de evolução entre diferentes linhagens.
À medida em que dois táxons divergem de um ancestral comum, os dois passam a acumular mutações em uma taxa constante e a diferir um do outro, fazendo com que o número de alterações seja proporcional ao tempo decorrido.