
| Doença de Wilson | |
|---|---|
 | |
| Especialidade | endocrinologia |
| Classificação e recursos externos | |
| CID-10 | E83.0 |
| CID-9 | 275.1 |
| CID-11 | 468161208 |
| OMIM | 277900 |
| DiseasesDB | 14152 |
| MedlinePlus | 000785 |
| eMedicine | med/2413 neuro/570 ped/2441 |
| MeSH | D006527 |
|
| |
A doença de Wilson ou degeneração hepatolenticular é uma doença hereditária autossômica recessiva cuja principal característica é o acúmulo tóxico de cobre nos tecidos, principalmente cérebro e fígado, o que leva o portador a manifestar sintomas neuropsiquiátricos e de doença hepática. É tratada com medicamentos que reduzem a absorção de cobre (suplementos de zinco) ou removem seu excesso do corpo (quelantes), mas um transplante de fígado pode ser necessário nos estágios tardios.
A taxa de incidência é de 1 em 30.000 pessoas e os sintomas geralmente aparecem entre os 6 e 20 anos de idade, embora casos em pacientes mais idosos têm sido descritos. A enfermidade foi nomeada em homenagem a Samuel Alexander Kinnier Wilson, neurologista britânico que descreveu a doença em 1912.
Causa

A doença de Wilson é causada por mutações em um gene do cromossomo 13 que codifica a proteína ATP7B, uma enzima do tipo ATPase que tem a função de secretar o cobre na bile.
Sinais e sintomas
Os principais locais de acúmulo de cobre são o fígado e o cérebro, consequentemente, doença hepática e sintomas neuropsiquiátricos são os principais achados clínicos que levam ao diagnóstico. Os portadores com distúrbio hepático mais proeminente geralmente são crianças e adolescentes, o que permite um diagnóstico e cuidados médicos mais precoces, enquanto que os doentes com predominância de sintomas neurológicos e psiquiátricos, tendem a estar em seus vinte anos de idade ou mais no momento que procuram atendimento médico.
Hepáticos
Hepatite culminando em cirrose é a apresentação hepática mais comum, mas alguns pacientes apresentam falência hepática fulminante.
Cerca de 5% de todos os pacientes são diagnosticados somente quando desenvolvem insuficiência hepática aguda fulminante, muitas vezes no contexto de uma anemia hemolítica (anemia devido à destruição das células vermelhas do sangue). Esta insuficiência hepática se traduz por anormalidades na produção de proteínas (identificada pela redução de fatores de coagulação) e prejuízo ao metabolismo das proteínas no fígado. A dificuldade de metabolização leva ao acúmulo de substâncias tóxicas, como a amônia, na corrente sanguínea. Esses resíduos irritam o cérebro, e o paciente desenvolve encefalopatia hepática (confusão mental, coma, convulsões e hipertensão intracaniana por edema).
Neuropsiquiátricos
Os fenômenos neuropsiquiátricos são demência, psicose e sinais de asteríxis (movimentos anormais, especialmente das mãos) e parkinsonismo (tremores mais evidentes em movimentos finos e lentos).
O quadro clínico inicial mais comum é o do paciente que fala de modo "arrastado" como se estivesse embriagado,e tem marcha de bases alargadas e titubeante, muito embora possa ter domínio perfeito de suas faculdades mentais. Posteriormente, a doença evolui para acometimento da percepção e da cognição (falta de lucidez).
Oculares
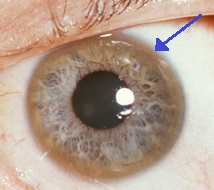
O acúmulo de cobre provoca uma mudança na pigmentação dos olhos, na membrana de Descemet, onde é possível verificar o aparecimento de anéis normalmente escuro-avermelhados (podendo apresentar colorações amarelo-esverdeada, marrom-esverdeada, amarelo-dourada ou marrom-dourada) ao redor da íris chamados de anéis de Kayser-Fleischer. A presença dos anéis de Kayser-Fleischer configura uma manifestação oftalmológica característica de pacientes com Doença de Wilson, mas apenas 2 em cada 3 pacientes com Wilson apresentam esse sintoma. A doença de Wilson também está associada a "catarata girassol", uma pigmentação marrom ou verde da cápsula anterior e posterior da lente do olho que causa uma lenta e progressiva redução da visão.
Outros órgãos
Também estão presentes problemas renais (nefrolitíase), oftálmicos (anel de Kayser-Fleischer, catarata), cardíacos (arritmia) e dermatológicos. A anemia por hemólise pode ocorrer em casos severos.
Diagnóstico
Os níveis de ceruloplasmina são anormalmente baixos (<0,2 g/L) em 80-95% dos casos. Níveis elevados de cobre na urina 24h, 100 μg/24h (ou 1.6 μmol/24h), confirmam intoxicação por cobre. A biópsia de fígado indicando esteatose ou cirrose é o exame mais sensível.
Tratamento
Em geral, recomenda-se uma dieta com baixo teor de alimentos ricos em cobre como cogumelos, nozes, chocolate, frutas secas, fígado e mariscos. Suplementos de zinco reduzem a absorção do cobre.
Geralmente, a D-penicilamina é o primeiro tratamento utilizado. Ela faz quelação do cobre para permitir sua excreção na urina. O monitoramento da quantidade de cobre na urina pode ser feito para garantir que a dose foi suficiente. Porém, em cerca de 20% experimentam parecem efeitos colaterais ou complicação do tratamento com penicilamina, tais como Lúpus eritematoso induzido por drogas (causando dores nas articulações e erupção cutânea) ou miastenia (fraqueza muscular). Naqueles que apresentaram sintomas neurológicos progressivamente piores, deve-se descontinuar o tratamento e usar outro quelante como cloridrato de trientina ou dimercaprol IM.
Em caso de insuficiência hepática severa, um transplante de fígado é a única cura efetiva.