
| Doença de Addison | |
|---|---|
 | |
| Especialidade | endocrinologia |
| Classificação e recursos externos | |
| CID-10 | E27.1-E27.2 |
| CID-9 | 255.4 |
| CID-11 | 1920929898 |
| OMIM | 103230 |
| DiseasesDB | 222 |
| MedlinePlus | 000378 |
| eMedicine | med/42 |
| MeSH | D000224 |
|
| |
A doença de Addison (DA), também conhecida como insuficiência adrenal primária e insuficiência adrenocortical primária crônica, é uma endocrinopatia potencialmente fatal caracterizada pela produção insuficiente de hormônios esteroides pelas glândulas adrenais. Os sintomas, geralmente graduais e progressivos, incluem hiperpigmentação de pele e mucosas, fraqueza, perda de peso, hipotensão postural e dor abdominal. Sem tratamento, a doença pode evoluir para uma crise adrenal, cursando com hipotensão arterial, vômitos, lombalgia e perda de consciência. Este quadro agudo pode ser desencadeado por stress por trauma físico, procedimentos cirúrgicos ou infecções.
A doença é causada pela destruição das células do córtex adrenal e a consequente deficiência na produção de cortisol e aldosterona. Esses danos celulares podem ser provocados por fatores autoimunes, complicações secundárias à infecção por tuberculose, fungos ou vírus, infiltração por neoplasias primárias ou metastáticas, hemorragia ou trombose adrenal, amiloidose, sarcoidose, hemocromatose ou terapia medicamentosa. Quando a doença é causada pela deficiência na produção de ACTH (produzido pela hipófise) ou de CRH (produzido pelo hipotálamo), é denominada insuficiência adrenal secundária. Apesar dessa distinção, a crise adrenal pode acontecer em todas as formas de insuficiência adrenal. A doença de Addison é geralmente diagnosticada por exames laboratoriais e de imagem. O tratamento exige acompanhamento médico regular e consiste basicamente na terapia de reposição hormonal, com a administração exógena de corticosteroides, como a hidrocortisona e fludrocortisona, por toda a vida.
A prevalência da doença de Addison no mundo é de cerca de 0,9 a 1,4 afetados em cada grupo de 10 000 pessoas e ocorre mais frequentemente em mulheres de meia idade. Normalmente, o prognóstico sob tratamento é bom a longo prazo. A doença foi descrita em 1855 por Thomas Addison, médico britânico graduado pela Escola de Medicina da Universidade de Edimburgo. O termo "addisoniano" é usado para descrever características da doença e seus portadores.
Causas
Em países desenvolvidos, estima-se que 68 a 94% dos casos de doença de Addison seja autoimune, causados por auto-anticorpos dirigidos às células adrenais contendo 21-hidroxilase, uma enzima envolvida na produção de cortisol e aldosterona.
O restante dos casos são devidos a tuberculose, aids, sarcoidose, amiloidose, hemocromatose, câncer metastático para as glândulas adrenais, hemorragia adrenal, síndrome de Waterhouse-Friderichsen (hemorragia maciça, geralmente bilateral, das adrenais causada por meningococcemia fulminante) e hiperplasia adrenal congênita.
A doença de Addison pode ser uma manifestação de uma síndrome poliendócrina autoimune quando há outras reações autoimunes contra outros órgãos. Assim, pode estar associada a hipotireoidismo, diabetes mellitus tipo 1, vitiligo, alopécia ou doença celíaca.
Sinais e sintomas
Mais comuns

A doença de Addison progride lentamente, e os sintomas podem ser discretos ou ausentes até que ocorra uma situação de stress. Os sintomas mais comuns são:
- Pressão arterial baixa que piora ao se levantar (hipotensão ortostática)
- Cansaço crônica com piora progressiva
- Vertigem (tontura especialmente ao ficar de pé e andar)
- Fraqueza muscular
- Artralgia e mialgia (dor muscular e nas articulações)
- Febre
- Anorexia
- Perda de peso
- Náusea e vômitos
- Diarreia
- Suor excessivo
- Dor de cabeça
- Irritação nos olhos
- Depressão, ansiedade e mudanças de humor
- Hipoglicemia (mais severa em crianças)
- Nas mulheres, o ciclo menstrual torna-se irregular ou ausente
- Tetania (particularmente após tomar leite) devido ao excesso de fosfato
- Adormecimento das extremidades, algumas vezes com paralisia dos mesmos, devido ao excesso de potássio
- Eosinofilia (excesso de eosinócitos)
- Poliúria (urinar em excesso)
- Desejo por sal e alimentos salgados, causado pela perda de sódio através da urina
- Déficit de atenção, confusão mental.
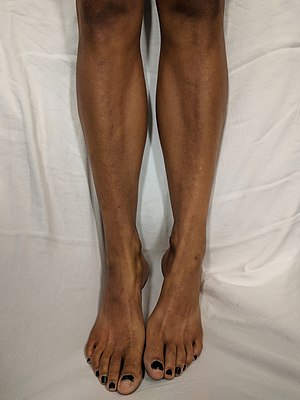
- Hiperpigmentação cutânea (escurecimento da pele e lábios) causada pela elevação das concentrações plasmáticas de ACTH (hormônio adrenocorticotrófico), que apresenta afinidade pelo receptor MC1 na pele, nas áreas expostas ao sol, com melasma suprarrenal (escurecimento) nos pontos de fricção, dobras cutâneas, linhas das palmas das mãos, genitália, cicatrizes recentes e em torno dos lábios.
Crise addisoniana
Uma situação de stress, outra doença ou acidente pode agravar a insuficiência adrenal e causar uma crise addisoniana. No entanto, a causa mais comum é a interrupção abrupta do uso de corticosteroide exógeno. Os sintomas podem incluir:
- Coloração marrom na língua e dentes devido ao acúmulo de ferro por hemólise
- Dor súbita e penetrante nas pernas, região lombar ou abdome
- Vômitos e diarreia severos, resultando em desidratação
- Hipotensão que pode ser severa, levando a choque hemodinâmico
- Coma
- Hipoglicemia
- Perda de memória
- A fraqueza progride até que o paciente se sinta continuamente fatigado, necessitando de repouso ao leito.
- Sua voz pode falhar, de modo que a fala se torna finalmente inaudível e indistinta.
Se não tratada rapidamente, uma crise addisoniana pode ser fatal. É uma emergência médica.
Diagnóstico
Em casos suspeitos de doença de Addison, é necessário demonstrar que o nível sanguíneo dos hormônios adrenais está baixo (geralmente medidos às 08:00, quando o seu nível é maior devido ao ciclo circadiano) e o nível de ACTH está elevado (refletindo que há estímulo da glândula hipofisária para a adrenal, mas que esse não produz hormônios suficientes). Além disso, pode-se dosar o nível dos hormônios adrenais, que permanecerão baixos após o estímulo da glândula com hormônio hipofisário sintético.
Além do diagnóstico da insuficiência adrenal, é necessário investigar a sua causa. A doença auto-imune pode ser investigada com a pesquisa de anticorpos para 21-hidroxilase. Se negativos, são necessários exames específicos para cada suspeita, incluindo exames de imagem da glândula adrenal. Como no Brasil a tuberculose, é uma das principais causas, muitos casos aonde a investigação não encontra uma etiologia são tratados como tuberculose.
Os números em que é classificado, no CID-10, a Insuficiência Adrenocortical Primária é E27.1, a Crise Addisoniana é E27.2 e Insuficiência adrenocortical induzida por drogas é E27.3, .
Prevalência
No Brasil cerca de 39% dos casos é de origem autoimune, 28% dos casos seja por paracoccidioidomicose, 11% por tuberculose, 7% por adrenoleucodistrofia e 15% por outras causas. Cerca de 40% dos pacientes com a forma autoimune da doença apresentam outras doenças autoimunes associadas. É mais comum em mulheres e crianças, sem apresentar predileção por etnia. Acontece principalmente na faixa dos 30 a 50 anos de idade, sendo especialmente mais precoce em pacientes com síndrome poliglandular autoimune .
Tratamento
O tratamento envolve a reposição de glicocorticoides como cortisol e, se necessário, de fluorohidrocortisona (0,05 a 0,15mg por dia) para reposição de aldosterona, além do tratamento da causa de base, que em alguns casos pode reverter a insuficiência adrenal.
Atualmente os pacientes costumam ser tratados com 15 a 25mg/dia de hidrocortisona, divididos em duas doses ou três doses por dia. Quando não há disponibilidade de hidrocortisona oral, pode-se utilizar 5 a 10mg de prednisona por dia ou dose equivalente de outro glicocorticoide como acetato de cortisona ou prednisolona.</ref name=silva>
Bibliografia
- Brandão Neto, R.A.; Carvalho, J.F (2014). «Diagnosis and classification of Addison's disease (autoimmune adrenalitis)». Autoimmunity Reviews (em inglês). 13 (4-5): 408-411. ISSN 1568-9972. Consultado em 4 de setembro de 2016
- Longmore, M.; Wilkinson, I.B.; Rajagopalan, S.R (2006). «Capítulo 9: Endocrinology: Addison's disease (adrenal insufficiency)». Mini Oxford Handbook of Clinical Medicine (em inglês) 6ª ed. Oxford: Oxford University Press. 312 páginas. ISBN 978-0-19-857071-4. Consultado em 4 de setembro de 2016
- Peterson, P.; Husebye, E.S (2014). «Capítulo 43: Polyendocrine Syndromes». In: Rose, N.R.; Mackay, I.R. The Autoimmune Diseases (em inglês) 5ª ed. San Diego: Elsevier Science. 605 páginas. ISBN 978-0-12-384929-8. Consultado em 4 de setembro de 2016
- Sahdev, A.; Reznek, R.H (2015). «Capítulo 44: Adrenal Imaging». In: Adam, A.; Dixon, A.K.; Gillard, J.H.; Schaefer-Prokop, C.M. Grainger & Allison's Diagnostic Radiology (em inglês). volume 1 6ª ed. Londres: Churchill Livingstone. 1031 páginas. ISBN 978-0-7020-6128-8. Consultado em 3 de setembro de 2016
- Silva, R.C.; Kater, C.E (dezembro de 1998). «Doença de Addison de etiologia auto-imune» (PDF). Arquivos Brasileiros de Endocrinologia e Metabologia. 42 (6): 431-443. ISSN 1677-9487. Consultado em 3 de setembro de 2016
- Silva, R.C.; Castro, M.; Kater, C.E.; Cunha, A.A.; Moraes, A.M.; Alvarenga, D.B.; Moreira, A.C.; Elias, L.L.K (outubro de 2004). «Insuficiência Adrenal Primária no Adulto: 150 Anos depois de Addison» (PDF). Arquivos Brasileiros de Endocrinologia e Metabologia. 48 (5): 724-738. ISSN 1677-9487. Consultado em 3 de setembro de 2016
- Ten, S.; New, M.; MacLaren, N (julho de 2001). «Clinical review 130: Addison's disease 2001» (PDF). The Journal of Clinical Endocrinology & Metabolism (em inglês). 86 (7): 2909-2922. ISSN 1945-7197. Consultado em 4 de setembro de 2016
Ver também
Ligações externas
- Hipofunção das glândulas supra-renais. Manual Merck. Fundação Merck Sharp & Dohme.
- «A menina que pode - literalmente - morrer de susto!»
- «Neuroimmunology, The Medical School, Birmingham University». - Abid R Karim, Birmingham UK
- «Addison's Disease Research Today»
- «Addison's Disease Self Help Group (ADSHG)». - grupo de suporte britânico
- «Addison Disease Orphanet»