
| Biopython | |
|---|---|
| Surgido em | 2000 (22–23 anos) |
| Última versão | 1.70 (11 de julho de 2017) |
| Criado por | Chapman B, Chang J |
| Página oficial |
www |
Biopython é uma biblioteca ou uma suite de ferramentas escritas em Python para manipulação de dados biológicos. Biopython apresenta uma coleção de classes, módulos e pacotes para análises de sequências biológicas, alinhamentos de sequências, estruturas de proteínas, genética de populações, filogenia, visualização de dados biológicos, detecção de regiões motivo em sequências, aprendizado de máquina, além de prover fácil acesso a bancos de dados biológicos.
O Projeto Biopython faz parte da coleção de Bio* projetos open-source mantidos pela Open Bioinformatics Foundation.
História

O desenvolvimento do Biopython iniciou-se no ano de 1999. Foi lançado em julho do ano 2000, entretanto a primeira versão surgiu apenas em março de 2001 e a primeira versão estável em dezembro de 2002.
A ideia de produzir um pacote de programas para análises biológicas usando a linguagem Python foi encabeçada por Jeff Chang e Andrew Dalke, inspirados por Ewan Birney e pelo projeto BioPerl. Além de Jeff e Andrew, Brad Chapman logo se tornou um dos grandes colaboradores do projeto. Atualmente, Biopython conta com centenas de contribuidores.
Desde o ano de 2012, Biopython superou BioPerl e passou a ser considerado o mais popular dos Bio* Projetos mantidos pela Open Bioinformatics Foundation.
Características
A utilização da linguagem Python foi sugerida por ser uma linguagem de alto nível, com tipagem dinâmica e com baixa curva de aprendizado, que vem sendo bastante adotada em projetos no ensino de princípios da computação. Biopython é um orientado a objetos. Biopython foi criado originalmente para rodar com Python 2, entretanto, a partir da versão 1.62 passou a suportar a execução em Python 3. A versão mais recente é a 1.70.
Biopython permite acesso a programas usados em bioinformática, manipulação de arquivos de diversos formatos, além de acesso remoto a diversas bases de dados. Dentre os programas, formatos e bases de dados, pode-se citar: SwissProt, UniGene, SCOP, ExPASy, GenBank, Medline, PubMed, além de execução de programas como BLAST (local ou remoto), Clustalw e FASTA.
Download e instalação
Biopython pode ser obtido em sua página oficial. Ele é suportado nos principais sistemas operacionais: Windows, Linux e MacOS. A instalação padrão em distribuições Linux requer apenas o download do arquivo de instalação, a descompactação e execução de três linhas de comando no terminal:
python setup.py build
python setup.py test
sudo python setup.py install
No Windows, é possível realizar a instalação através de arquivos executáveis ou por meio do pip. Para isso acesse o CMD e execute o comando abaixo (o endereço do pip pode variar de acordo com a versão instalada do Python):
C:\Python27\Scripts\pip install biopython
Instalando no MacOS:
pip install biopython
Para outros sistemas operacionais ou outros métodos de instalação, consulte a documentação oficial do Biopython.
Exemplos
Manipulações de sequências
Em Biopython, sequências são declaradas como objetos e não como strings. O módulo necessário para manipulação de sequências é o Bio.Seq. Para trabalhar com sequências de nucleotídeos e aminoácidos é necessário ainda declarar o módulo Bio.Alphabet e a classe IUPAC, que define padrões internacionais para os alfabetos utilizados tanto para sequências de DNA, RNA e proteínas.

A classe Seq apresenta métodos para manipulação de sequências, como por exemplo, a detecção da sequência complementar e complementar reversa de uma sequência de nucleotídeos pertencentes a DNA, métodos para transcrição (DNA -> RNA) e transcrição reversa (RNA -> DNA), além de métodos para tradução (RNA -> proteína / DNA -> proteína)..
from Bio.Seq import Seq
from Bio.Alphabet import IUPAC
# Criando uma sequencia exemplo
seq_exemplo = Seq("ATG")
# Sequencia reversa e reversa complementar
seq_complementar = seq_exemplo.complement() #TAC
seq_complementar_reversa = seq_exemplo.reverse_complement() #CAT
# Transcricao
seq_rna = seq_exemplo.transcribe() #AUG
seq_dna = seq_rna.back_transcribe() #ATG
# Traducao
seq_proteina_rna = seq_rna.translate() #M
seq_proteina_dna = seq_dna.translate() #M
Manipulando arquivos no formato FASTA
O formato FASTA é um padrão bastante utilizado em aplicações de bioinformática. Ele consiste num arquivo de sequências, a qual cada sequência possui um cabeçalho iniciado com o sinal de ">". Veja abaixo um exemplo de arquivo FASTA:
>Titulo_cabecalho_sequencia
CGATCGATCGACTATCAGCATCGATCGACTAGCATCGACTACGATGA
GACTATCAGCATCGATCGACTAGCATCGACTACGACGATCGATCTGA
CGACTATCAGCATCGATCGACTAGCATCGACTACGATGACGATCGAT
CGATCGAT
Biopython apresenta o módulo SeqIO para leitura e manipulação de arquivos em diversos formatos. Por exemplo, dado um arquivo denominado "nome_arquivo.fasta", pode-se utilizar o método SeqIO.parse para percorrer o arquivo e obter informações como: título do cabeçalho e sequência completa.
from Bio import SeqIO
for fasta in SeqIO.parse("nome_arquivo.fasta","fasta"):
#imprime id do cabecalho
print fasta.id
#imprime sequencia completa
print fasta.seq
O módulo SeqIO também pode ser utilizado para manipular outros formatos, como o formato GenBank, que armazena informações de anotações de proteínas em genomas.
from Bio import SeqIO
# Manipulando arquivos no formato genbank
genomas = SeqIO.parse('arquivo.genbank', 'genbank')
# Convertendo no formato FASTA
for genoma in genomas:
SeqIO.write(genoma, genoma.id + '.fasta', 'fasta')
Anotação de sequências
A classe SeqRecord do módulo SeqIO permite o acesso a sequências de arquivos de anotação (formato "genbank"), além de outras informações como nome, descrição, informações sobre genes, CDS (sequência codificante), regiões de repetição, dentre outras informações.
#-*- Coding: utf-8 -*-
from Bio import SeqIO
arquivo_gb = SeqIO.read('pTC2.gb', 'genbank')
arquivo_gb.name
# 'NC_019375'
arquivo_gb.description
# 'Providencia stuartii plasmid pTC2, complete sequence.'
arquivo_gb.features[14]
# SeqFeature(FeatureLocation(ExactPosition(4516), ExactPosition(5336), strand=1), type='mobile_element')
arquivo_gb.seq
# Seq('GGATTGAATATAACCGACGTGACTGTTACATTTAGGTGGCTAAACCCGTCAAGC...GCC', IUPACAmbiguousDNA())
Estruturas de proteínas

Biopython possui pacotes para manipulação de arquivos de estruturas de proteínas (formato PDB). O módulo Bio.PDB foi desenvolvido por Thomas Hamelryck e Bernard Manderick. Este módulo requer o pacote NumPy.
Bio.PDB converte um arquivo de estrutura tridimensional em um objeto Structure seguindo a chamada arquitetura EMCRA (Estrutura, Modelo, Cadeia, Resíduo e Átomo; no original SMCRA architecture)[1]. Nesta abordagem:
- uma estrutura consiste em modelos;
- um modelo consiste em cadeias;
- uma cadeia consiste em resíduos;
- um resíduo consiste em átomos.
Estrutura (structure)
O objeto structure está no topo da hierarquia. Seu id é uma string dada pelo usuário. Ela pode ser lida a partir do método get_structure( ), que deve ser aplicado a um objeto de PDBParser( ).
from Bio.PDB import *
parser = PDBParser()
estrutura = parser.get_structure('NOME', 'FILE.pdb')
Modelo (model)
Estruturas obtidas por cristalografia de raio-x tendem a apresentar apenas um modelo, enquanto estruturas obtidas por NMR podem apresentar diversos modelos.
from Bio.PDB import *
# Declaracoes do objeto
parser = PDBParser()
estrutura = parser.get_structure('NOME', 'FILE.pdb')
primeira_estrutura = estrutura[0]
Cadeia (chain)
Cadeias representam a estrutura quaternária de uma proteína, que pode ter uma ou mais. Para Biopython, o id de uma cadeia, em geral, é um caracter.
from Bio.PDB import *
parser = PDBParser()
estrutura = parser.get_structure('NOME', 'FILE.pdb')
# Coletando a primeira cadeia
primeira_estrutura = estrutura[0]
cadeia_A = primeira_estrutura['A']
Resíduo (residue)
O id de um resíduo é uma tupla composta por três elementos:
- hetero-field (hetfield): "W" em caso de molécula de água; "H_" seguido de um nome de resíduo (ex.: "H_ALA"); e vazio (padrão para qualquer aminoácido);
- sequence identifier (resseq; identificador de sequência): um valor inteiro que descreve a posição do resíduo;
- insertion code (icode; código de inserção): usado para preservar posições de resíduos; esse campo recebe uma string (ex.: 'A).
Entretanto, o hetero-field e o insertion code são opcionais. Observe o exemplo a seguir, a qual o 100º resíduo é extraído da cadeia A de uma proteína com apenas um modelo:
from Bio.PDB import *
parser = PDBParser()
estrutura = parser.get_structure('NOME', 'FILE.pdb')
# Coletando o residuo 100
primeira_estrutura = estrutura[0]
cadeia_A = primeira_estrutura['A']
residuo_100 = cadeia_A[100]
Dois método podem ser aplicados a resíduos:
| Método | Retorna |
| get_resname( ) | nome do resíduo |
| has_id(name) | testa se há certo átomo |
Átomo (atom)
O id de um átomo é uma string única para um resíduo (ex.: 'CA'; carbono-alfa).
from Bio.PDB import *
parser = PDBParser()
estrutura = parser.get_structure('NOME', 'FILE.pdb')
# Coletando o carbono-alfa do residuo 100
primeira_estrutura = estrutura[0]
cadeia_A = primeira_estrutura['A']
residuo_100 = cadeia_A[100]
ca_residuo_100 = residuo_100['CA']
Além disso, Biopython permite a aplicação de diversos métodos para obtenção de informações sobre átomos.
| Método | Retorna |
| get_name() | nome do atomo |
| get_id() | id |
| get_coord() | coordenadas atomicas |
| get_vector() | coordenadas como vetor |
| get_bfactor() | fator B |
| get_occupancy() | ocupancia |
| get_altloc() | localizacao alternativa |
| get_sigatm() | parametros atomicos |
| get_anisou() | fator B anisotropico |
| get_fullname() | nome completo do atomo |
Baixando arquivos PDB
É possível fazer o download de arquivos PDB usando o método retrieve_pdb_file( ), que recebe como parâmetro o código PDB.
from Bio.PDB import *
pdb = PDBList()
pdb.retrieve_pdb_file('1BGA')
Salvando arquivos no formato PDB
Bioptyhon fornece a classe PDBIO( ) para permitir o salvamento de arquivos no formato PDB. No exemplo a seguir é criado um arquivo contendo apenas "alaninas":
class AlaSelect(Select):
def accept_residue(self, residue):
if residue.get_name()=='ALA':
return True
else:
return False
io = PDBIO()
io.set_structure(s)
io.save('somente_alaninas.pdb', AlaSelect())
Cálculo de distância entre átomos
No exemplo a seguir é demonstrado o cálculo de distância entre dois átomos (posições 1 e 2) da cadeia A de uma estrutura tridimensional armazenada no arquivo "FILE.pdb":
from Bio.PDB import *
# Declaracoes do objeto
parser = PDBParser()
estrutura = parser.get_structure('NOME', 'FILE.pdb')
# Coletando carbonos alfa de dois residuos da cadeia A: 1 e 2
ca_residuo_1_A = estrutura[0]['A'][1]['CA']
ca_residuo_2_A = estrutura[0]['A'][2]['CA']
distancia = ca_residuo_1_A - ca_residuo_2_A
print distancia # 4.879
Salvando um arquivo PDB em formato FASTA
É possível salvar um arquivo PDB em formato FASTA, entretanto é necessário o uso de CaPPBuilder( ) para auxiliar nesse processo.
from Bio.PDB import *
parser = PDBParser()
peptideos = CaPPBuilder()
estrutura = parser.get_structure('BGA', '1BGA.pdb')
# Criando um arquivo FASTA
w = open("1BGA.fasta","w")
# Loop que le todas as cadeias de uma estrutura de proteina
for cadeia in estrutura[0]:
cadeia_atual = cadeia.id
# Para cada cadeia leia todos os residuos
for residuo in peptideos.build_peptides(cadeia):
residuo_atual = residuo.get_sequence()
fasta = ">Cadeia_%s\n%s\n" %(cadeia_atual,residuo_atual)
print fasta
w.write(fasta)
w.close()
Alinhamento estrutural
O módulo Bio.PDB permite o alinhamento estrutural entre arquivos PDB usando a classe Superimposer( ). Alinhamento estrutural é uma técnica da bioinformática estrutural que visa alinhar (rotacionar e translacionar) uma ou mais estruturas tridimensionais de macromoléculas a uma macromolécula referência.
No exemplo a seguir, o arquivo "2.pdb" (PDB2) será alinhado estruturalmente ao arquivo "1.pdb" (PDB1). Ao final, um novo arquivo denominado "3.pdb", com novas coordenadas será salvo no mesmo diretório de execução do arquivo. Observe que para o alinhamento ser realizado, é necessário percorrer a variável que recebe a estrutura da proteína para obter-se os átomos. Por fim, os átomos do PDB2 são alinhados aos átomos do PDB1 e um novo arquivo é salvo com as novas coordenadas e o RMS é exibido.
from Bio.PDB import *
import numpy
# Lendo os pdbs
PDB1 = PDBParser().get_structure('PDB1','1.pdb')
PDB2 = PDBParser().get_structure('PDB2','2.pdb')
# Extraindo os atomos
ref_atoms = [] #PDB1
alt_atoms = [] #PDB2
for ref_chain, alt_chain in zip(PDB1[0], PDB2[0]):
for ref_res, alt_res in zip(ref_chain, alt_chain):
for ref_a, alt_a in zip(ref_res, alt_res):
ref_atoms.append(ref_a)
alt_atoms.append(alt_a)
# Fazendo a sobreposicao
si = Superimposer()
si.set_atoms(ref_atoms, alt_atoms)
si.apply(PDB2.get_atoms())
# Salvando o novo arquivo PDB
io = PDBIO()
io.set_structure(PDB2)
io.save("3.pdb")
# Obtendo o RMS
print si.rms
Visualização de dados biológicos

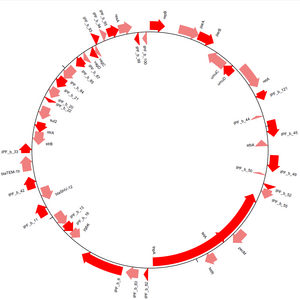
Biopython possui um conjunto de métodos para auxiliar na produção de visualização de dados biológicos. Python apresenta o módulo pylab, que auxilia na construção de visualizações de dados. Biopython apresenta o pacote Bio.Graphics que trabalha em paralelo com a biblioteca ReportLab.
Visualização de genomas
O pacote Bio.Grafics apresenta o módulo GenomeDiagram para visualização de sequências. Bio.Graphics pode ser utilizado para visualização de regiões codificadoras e comparações sintênicas entre genomas. No exemplo a seguir temos a implementação da visualização de CDS em um arquivo de anotações gênicas no formato GenBank.
from Bio import SeqIO
from Bio.Graphics import GenomeDiagram
from reportlab.lib import colors
from reportlab.lib.units import cm
# Recebendo o arquivo com as anotacoes genicas
arquivo = SeqIO.read("arquivo.gbk", "genbank")
# Declarando o diagrama
gd_diagram = GenomeDiagram.Diagram("plasmideo")
gd_track_for_features = gd_diagram.new_track(1, name="Anotacoes")
gd_feature_set = gd_track_for_features.new_set()
# Crie uma retangulo para cada gene
for feature in arquivo.features:
if feature.type != "gene":
# Despreza a feature que nao sao genes
continue
if len(gd_feature_set) % 2 == 0:
color = colors.HexColor('#79B134')
else:
color = colors.HexColor('#8DE91D')
gd_feature_set.add_feature(feature, color=color, label=True)
# Desenhando
gd_diagram.draw(format="circular", circular=True, pagesize=(25*cm,25*cm),start=0, end=len(arquivo), circle_core=0.7)
# Salvando figura
gd_diagram.write("plasmideo.png", "PNG")
Sintenia entre genomas
Define-se sintenia como regiões conservadas entre diferentes genomas de dois ou mais organismos. Para determinação de sintenia, utiliza-se ferramentas de alinhamento local, como por exemplo o BLAST. Em bioinformática, a visualização de sintenia permite comparações entre grandes blocos de genomas e pode ser realizada com ferramentas como ACT, CONTIGuator, MUMmer.
No exemplo a seguir a sintenia entre dois genomas é executada. Como entrada são utilizados dois arquivos no formato GenBank.
# -*- coding: utf-8 -*-
# Sintenia entre genomas
# Pacotes necessarios
from Bio import SeqIO
from Bio.Graphics import GenomeDiagram
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics.GenomeDiagram import CrossLink
from reportlab.lib import colors
from Bio.Blast.Applications import *
import sys
# Recebendo os dados pela chamada do programa: python script.py A B
A = sys.argv[1]
B = sys.argv[2]
A1 = SeqIO.read(A,"genbank")
B1 = SeqIO.read(B,"genbank")
# Usando BLAST para comparar genomas
SeqIO.convert(A, "genbank", A+".fasta", "fasta")
SeqIO.convert(B, "genbank", B+".fasta", "fasta")
comando_blastn = NcbiblastnCommandline( \
query=A+".fasta", subject=B+".fasta", \
outfmt="'6 qstart qend sstart send pident'",\
out="blast_"+A+"_"+B+".txt")
stdout, stderr = comando_blastn()
blast = open("blast_"+A+"_"+B+".txt")
# Iniciando a figura
name = A+"_"+B
gd = GenomeDiagram.Diagram(name)
gA = gd.new_track(1,name="A",height=0.5, \
start=0,end=len(A1))
gA1 = gA.new_set()
gB = gd.new_track(3,name="B",height=0.5, \
start=0,end=len(B1))
gB1 = gB.new_set()
# Cores CDSs L intercalado
c1 = "#79B134"
c2 = "#8DE91D"
# Colore um quadrado para cada CDS do arquivo A
cont = 1
for i in A1.features:
if i.type == "CDS":
if cont % 2 == 1:
color_atual = c1
else:
color_atual = c2
gA1.add_feature(i, label=False, \
label_position="start",color=color_atual)
cont += 1
if i.type == "rRNA":
color_atual = colors.blue
gA1.add_feature(i, label=False,\
label_position="start",color=color_atual)
# Colore um quadrado para cada CDS do arquivo B
cont = 1
for i in B1.features:
if i.type == "CDS":
if cont % 2 == 1:
color_atual = c1
else:
color_atual = c2
gB1.add_feature(i, label=False,\
label_position="start",color=color_atual)
cont += 1
if i.type == "rRNA":
color_atual = colors.blue
gB1.add_feature(i, label=False,\
label_position="start",color=color_atual)
# Marca na figura os trechos sintenicos
for b in blast:
qstart = int(b.split("\t")[0])
qend = int(b.split("\t")[1])
sstart = int(b.split("\t")[2])
send = int(b.split("\t")[3])
identidade = (float(b.split("\t")[4])*0.8)/100
# Detectando inversoes
qinv = qend L qstart
sinv = send L sstart
if (qinv > 0 and sinv > 0) or \
(qinv < 0 and sinv < 0):
cor = colors.Color\
(1,.341176,.341176,identidade)
else:
cor = colors.firebrick
gd.cross_track_links.append(CrossLink((gA, \
qstart, qend),(gB, sstart, send),color=cor))
gd.draw(format="linear", pagesize=(8*cm,29.7*cm), \
fragments=1)
# Gera a figura de sintenia
gd.write(name + ".pdf", "PDF")
O primeiro genoma é exibido acima e o segundo abaixo. Linhas vermelhas indicam regiões sintênicas. Cada linha verde representa um gene. Linhas azuis representam regiões codificadoras de RNA ribossomal.

Filogenia


Biopython apresenta o módulo Bio.Phylo, que fornece métodos para manipular árvores filogenéticas. Bio.Phylo suporta diversos formatos, como por exemplo phyloXML, Newick e NEXUS. Manipulações e travessias de árvore comuns são suportadas através dos objetos "Tree" e "Clade". Os exemplos incluem a conversão e o agrupamento de arquivos de árvores, a extração de subconjuntos de uma árvore, a alteração da raiz de uma árvore e a análise de recursos de ramificações, como comprimento ou pontuação.
Árvores enraizadas podem ser desenhadas em ASCII ou usando o matplotlib, e a biblioteca Graphviz pode ser usada para criar layouts não-arraigados.
Genética de populações
O módulo Bio.PopGen adiciona suporte ao Biopython para Genepop, um pacote de software para análise estatística da genética de populações. Isso permite análises do equilíbrio de Hardy-Weinberg, desequilíbrio de ligação e outras características das frequências alélicas de uma população.
Este módulo também pode realizar simulações genéticas de populações usando a teoria coalescente com o programa fastsimcoal2.
Acessando bases de dados online
Biopython permite o download de arquivos de bases de dados online, como por exemplo as bases de dados de nucleotídeos do NCBI, por meio do módulo Entrez.
#-*- Coding: utf-8 -*-
# Download de genomas do NCBI e salvar em formato FASTA
from Bio import Entrez
from Bio import SeqIO
# Definições
saida = open('arquivo.fasta', "w")
Entrez.email = 'meu_email@examplo.com'
itens_para_baixar = ['FO834906.1', 'FO203501.1']
# Para cada download
for item in itens_para_baixar:
handle = Entrez.efetch(db='nucleotide', id=item, rettype='gb')
seqRecord = SeqIO.read(handle, format='gb')
handle.close()
saida.write(seqRecord.format('fasta'))
Wrappers
Biopython possui métodos para fácil execução de ferramentas externas e manipulação dos arquivos de saída. Os principais pacotes são:
- Bio.Align.Applications: permite acesso a ferramentas de alinhamento;
- Bio.Blast.Applications: permite acesso a alinhamentos locais de sequência com BLAST;
- Bio.Emboss.Applications: permite o acesso a aplicações Emboss;
- Bio.Sequencing.Applications: permite acesso a ferramentas de manipulação de dados de sequenciamento.
Alinhamentos múltiplos
O pacote Bio.Align permite fácil acesso a ferramentas de alinhamento múltiplos de sequências executadas por linha de comando, dentre eles: Clustal Omega, Clustal W, DIALIGN 2-2, MAFFT, MUSCLE, PRANK, PROBCONS e T-Coffee.
| Classe | Ferramenta |
|---|---|
| MuscleCommandline | MUSCLE |
| ClustalwCommandline | Clustal W (versão 1 e 2) |
| ClustalOmegaCommandline | Clustal Omega |
| PrankCommandline | PRANK |
| MafftCommandline | MAFFT |
| DialignCommandline | DIALIGN2-2 |
| ProbconsCommandline | PROBCONS |
| TCoffeeCommandline | TCoffee alignment program |
| MSAProbsCommandline | MSAProbs |
A seguir será demonstrado um exemplo de alinhamento múltiplo usando o software Clustal W:
from Bio.Align.Applications import ClustalwCommandline
entrada = "arquivo.fasta"
clustalw_cline = ClustalwCommandline("clustalw2", infile=entrada)
print clustalw_cline
# clustalw2 -infile=arquivo.fasta
BLAST
BLAST é uma ferramenta para busca de alinhamentos locais de sequências. Biopython permite execução de BLAST localmente e pela internet.
BLAST pela Internet
Biopython permite a execução de BLAST pelo web service do NCBI usando a função qblast( ) da classe NCBIWWW do módulo Bio.Blast. A execução de BLAST online pela classe NCBIWWW requer como entrada a versão do BLAST utilizada para busca (blastn, blastp, blastx ou tblastn), além da base de dados, a sequência armazenada no objeto que recebeu o arquivo FASTA de busca e um tipo de formato para saída dos dados.
from Bio.Blast import NCBIWWW
from Bio import SeqIO
fasta = SeqIO.read("seq.fasta", format="fasta")
output = NCBIWWW.qblast("blastn", "nt", fasta.seq, format_type="Text")
print output.read()
BLAST Local
Executar BLAST localmente, em geral, é mais rápido que pela Internet. Além disso, isso permite que você construa suas próprias bases de dados. Entretanto, para isso é necessário:
- Suíte NCBI-BLAST+ instalada;
- Base de dados desejadas para consultas estejam armazenadas localmente.Você pode comparar duas sequências em localmente usando o comando NcbiblastnCommandline (usado para sequências de nucleotídeos). Para isso, é necessário que a suite de aplicações NCBI-BLAST+ esteja instalada.Biopython também permite a execução de outras versões do programa BLAST:
from Bio.Blast.Applications import * comando_blastn = NcbiblastnCommandline(query="seqA.fasta", subject="seqB.fasta", outfmt=0, out="output.txt") stdout, stderr = comando_blastn() resultado_blast = open("output.txt","r") linhas = resultado_blast.read() print linhas
| Método | Versão do BLAST |
|---|---|
| NcbiblastpCommandline( ) | blastp |
| NcbiblastxCommandline( ) | blastx |
| NcbitblastnCommandline( ) | tblastn |
| NcbitblastxCommandline( ) | tblastx |
| NcbiblastnCommandline( ) | blastn |