
| Angioedema hereditário | |
|---|---|
 | |
| Especialidade | hematologia |
| Classificação e recursos externos | |
| CID-10 | D84.1 (ILDS D84.110) |
| CID-9 | 277.6 |
| CID-11 | 795969334 |
| OMIM | 106100 |
| DiseasesDB | 1821 |
| MedlinePlus | 001456 |
| eMedicine | article/1048994 |
| MeSH | D054179 |
|
| |
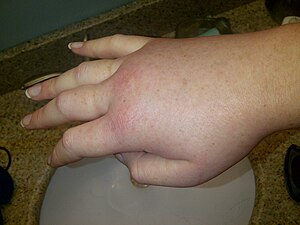
O angioedema hereditário (também conhecido pela sigla AEH) é uma doença rara do sistema imunitário, com herança autossômica dominante, que causa ataques episódicos de edema (inchaço) que podem afetar a face, as extremidades, a genitália, o trato gastrointestinal e as vias aéreas superiores. O edema da mucosa intestinal pode ocasionar vômitos e espasmos intestinais dolorosos semelhantes à cólica, mimetizando uma obstrução intestinal. O edema de vias aéreas pode apresentar risco de morte. Os episódios podem ser provocados por trauma, cirurgias, procedimentos dentários, menstruação, alguns medicamentos, doenças virais e estresse. Entretanto, eles não são prontamente determinados. Essa doença afeta aproximadamente uma a cada 10.000-50.000 pessoas.
A causa subjacente do AHE é atribuída à herança autossômica dominante de mutações no gene do inibidor da C1 esterase (C1-INH, gene SERPING1), que está localizado no cromossomo 11 (11q12-q13.1). Até o presente momento, há mais de 300 mutações genéticas conhecidas que resultam na deficiência funcional do inibidor da C1 esterase. A maior parte dos pacientes com AHE possuem história de familiares com o mesmo problema, entretanto, 25% resultam de novas mutações. O baixo nível de inibidores de C1 no plasma leva ao aumento da ativação das vias que liberam bradicinina, o composto químico responsável pelo angioedema (devido ao aumento da permeabilidade vascular) e pela dor observada em indivíduos com AHE.
A forma mais comum da doença é o AHE tipo I, que é resultado de níveis anormalmente baixos da enzima inibidora da C1 esterase, que faz parte do sistema complemento. Elas ajudam a regular várias funções do organismo (por exemplo, fluxo de fluidos corporais para dentro e fora das células). O AHE tipo II, que conta com 15-20% dos casos, é uma forma menos comum da doença, que ocorre como resultado da produção de proteínas do complemento anormais. O tipo III é uma forma muito rara recentemente documentada. Afeta predominantemente as mulheres e é influenciada pela exposição a estrógenos ou terapia de reposição hormonal (por exemplo, contraceptivos orais e na gestação) e não está associada à deficiência de C1-INH. O tipo III está ligado ao aumento da atividade da cininogenase, levando à elevação dos níveis de bradicinina. Alguns pacientes com o AHE tipo III têm mutação no gene F12, que produz uma proteína envolvida na coagulação sanguínea.
A doença foi descrita pelo Dr. Heinrich Quincke, em 1882.