
| Anemia falciforme | |
|---|---|
 | |
| Especialidade | Hematologia |
| Sintomas | Ataques de dor, anemia, inchaço das mãos e dos pés, infecções bacterianass, acidente vascular cerebral |
| Complicações | Dor crónica |
| Início habitual | 5–6 meses de idade |
| Causas | Genéticas |
| Método de diagnóstico | Análises ao sangue |
| Tratamento | Vacinação, antibióticos, beber líquidos, suplementos de ácido fólico, analgésicos, transfusões de sangue |
| Prognóstico | Esperança de vida de 40–60 anos (países desenvolvidos) |
| Frequência | 4,4 milhões (2015) |
| Mortes | 114 800 (2015) |
| Classificação e recursos externos | |
| CID-10 | D57 |
| CID-9 | 282.6 |
| CID-11 | 975559344 |
| OMIM | 603903 |
| DiseasesDB | 12069 |
| MedlinePlus | 000527 |
| eMedicine | med/2126 oph/490 ped/2096 emerg/26 emerg/406 |
| MeSH | D000755 |
| GeneReviews | |
|
| |
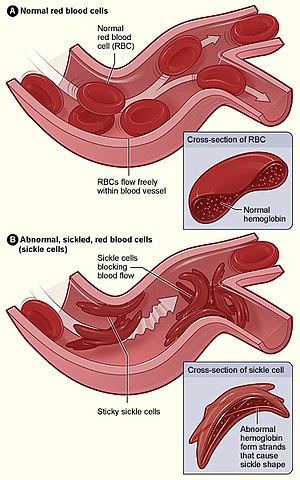
Anemia drepanocítica, drepanocitose ou anemia falciforme (do latim falci-, foice e -forme, formato de) é uma doença hematológica hereditária monogénica, caracterizada pela produção anormal de hemoglobinas, entre as quais a mais comum é a forma HbS (de Sickle, foice), que sob determinadas condições de desoxigenação, polimeriza, deformando as hemácias, que assumem uma forma semelhante a foices, causando deficiência no transporte de oxigénio e gás carbónico e outras complicações, nos indivíduos acometidos pela doença. Por esse motivo, a anemia drepanocítica é também conhecida por anemia falciforme. Do ponto de vista patogenético, está classificada entre as anemias por "defeito da síntese de hemoglobina", a proteína que transporta oxigénio presente nos glóbulos vermelhos, embora a anemia seja em parte determinada pela hemólise esplénica. Os sintomas geralmente começam a aparecer entre os 5 e 6 meses de idade. Podem desenvolver-se vários problemas de saúde, tais como crises de dor, anemia, infecções bacterianas e acidentes vasculares cerebrais. Podem desenvolver-se dores crónicas à medida que a pessoa envelhece. A esperança média de vida nos países desenvolvidos varia entre os 40 e 60 anos.
A anemia falciforme ocorre quando a pessoa herda duas cópias anormais do gene de hemoglobina, um de cada progenitor. Existem vários subtipos, dependendo da mutação exacta em cada gene de hemoglobina. Um ataque pode configurar-se por mudanças de temperaturas, stress ou desidratação em sítios de elevada altitude. Uma pessoa com uma só cópia anómala normalmente não apresenta sintomas e diz-se que possui um traço drepanocítico ou falcémico. Estas pessoas são também referidas como portadoras da doença. O diagnóstico é feito por um exame de sangue e alguns países examinam todos os bebés quando nascem. O diagnóstico pode também ser feito durante a gravidez.
O tratamento da pessoa com anemia falciforme pode incluir a prevenção a infecções através da vacinação e uso de antibióticos, uma elevada ingestão de líquidos, a suplementação de ácido fólico e fármacos analgésicos. Outras medidas podem incluir a transfusão de sangue e a prescrição de hidroxicarbamida (hidroxiureia). Um reduzido percentual de pessoas pode ser curado mediante um transplante de células da medula óssea.
Em 2013, cerca de 3,2 milhões de pessoas sofriam de anemia drepanocítica, enquanto mais de 43 milhões apresentavam traço falciforme. Considera-se que cerca de 80% dos casos de anemia falciforme ocorram na África subsariana. É relativamente comum em algumas partes da Índia, da península arábica e entre pessoas de origem africana que vivem noutras parte do mundo. No mesmo ano, a anemia falciforme provocou 176.000 mortes, acima das 113.000 mortes registadas em 1990. A primeira descrição médica da doença foi realizada pelo médico norte-americano James B. Herrick, em 1910. Em 1949, E. A. Beet e J. V. Neel determinaram a origem genética. Em 1954 foi descrito o efeito protector contra a malária do traço falciforme.
Causas
A presença da anemia falciforme, é determinada por uma quantidade elevada de hemácias deformadas. Em indivíduos normais, as células de transporte de gases, hemácias (também conhecidas como eritrócitos ou glóbulos vermelhos), têm forma arredondada côncava e flexível, e possuem em si moléculas de hemoglobina,que são responsáveis por fazer as ligações gasosas. Essa constituição permite que essas células consigam executar sua função mesmo através dos mais finos capilares.
A formação dessa hemoglobina, determinada por um par genético no cromossomo 11, muda nos indivíduos falciformes. Neles, há a presença de ao menos um gene mutante, que leva o organismo a produzir a hemoglobina S (HbS).
A descoberta dos polimorfismos da mutação (GAT→GTG) no gene que codifica a cadeia β da hemoglobina, originando diferentes haplótipos da doença, permitiu um melhor e mais amplo conhecimento em torno da heterogeneidade clínica nos pacientes falcêmicos. Analisando a hemoglobina na sua estrutura normal e mutante, sua produção e evolução, pode-se ter um entendimento mais completo da fisiopatologia desta doença e da sua complexidade clínica. A substituição da base nitrogenada timina (T) por adenina (A), ocasionando a substituição do aminoácido ácido glutâmico por valina, na posição seis da cadeia β, é a mesma para todo paciente. A polimerização da hemoglobina S (HbS) e a falcização das hemácias são extremamente bem conhecidas. As variações das condições climáticas, sociais, econômicas e de cuidados médicos contribuem para esta diversidade, mas não explicam todo o seu contexto ( Nagel, R.L. et al. Hematologically and genetically distinct forms of sickle cell anemia in Africa. N. Engl. J. Med., 312: 880-4, 1985, Aspectos moleculares da anemia falciforme Galiza Neto & Pitombeira.)
A homozigosidade para esta mutação é a causa dessa anemia falciforme. Um heterozigoto tem uma mistura dos dois tipos de hemoglobinas, A (HbA) e S (HbS), além de um tetrâmero híbrido de Hemoglobina. Ela consegue transportar o oxigênio mas, quando o mesmo passa para os tecidos, as moléculas da sua hemoglobina se aglutinam em formas gelatinosas de polímeros, também chamadas tactóides, que acabam por distorcer as hemácias, que se tornam duras e quebradiças devido às mudanças na sua membrana.
Quando recebem novamente o oxigênio, podem ou não reganhar seu formato: após algum tempo, por não suportar bem modificações físicas, a hemoglobina pode manter a forma gelatinosa permanentemente e, consequentemente, a deformação que ela gera. Nessa forma, sua vida útil se extingue mais rapidamente, o que pode vir a causar anemia hemolítica (ou comum). Contudo, ao contrário da anemia comum, não há tratamento definitivo para a forma falciforme. O gene causador desse último problema tem uma relação de co-dominância com o gene normal. Assim, há indivíduos portadores de uma forma branda e de uma forma severa da mesma doença.
Doença falciforme
A anemia falciforme obedece a um modelo de herança tipo autossômica recessiva É chamado portador de anemia falciforme o paciente que apresenta o gene da hemoglobina S (HbS) em homozigose e portador do traço falcêmico o indivíduo que apresenta o gene da HbS em heterozigose (HbS - HbA).
Observe-se porém que existe expressão clínica patológica caso o gene HbS se apresente em qualquer heterozigota mista, que não a combinação com a hemoglobina mais frequente (HbA) caracterizando o portador normal traço falcêmico. Na combinação do gene HBS com genes determinantes de variantes da hemoglobina (HbC; HbD) ou de hemoglobinas anômalas causadoras de outras formas de anemia hereditária tipo a talassemia Caso o gene HBS temos o que se denomina Doença Falciforme (SC; SD; S-talassemia, etc.) com sintomatologia semelhante, ou mais grave a da anemia falciforme.
Sinais e sintomas
Há a presença de alguns dos sintomas clássicos da anemia, causados pela falta e ineficiência de hemácias como:
- Fadiga (cansaço);
- Astenia (fraqueza);
- Palidez (principalmente nas conjuntivas e mucosas).
Há, contudo, a presença de uma gama de sintomas característicos da anemia falciforme aguda, que são causados pelo aumento da viscosidade sanguínea, que é a aglomeração de hemácias comprometidas. Por causa disso, pode haver formação de trombos (coágulos) nas mais diversas áreas do organismo, com défice do transporte sanguíneo para a área. Em regiões musculares ou conjuntivas, isso pode causar crises de dor intensa.
Concomitantemente a isso, há um aumento do número de hemácias comprometidas, uma vez que a acidose e a deficiência de oxigênio facilita a deformação permanente. Pode causar também hemorragia, descolamento retiniano, priapismo, acidente vascular cerebral, enfarte, calcificações em ossos com dores agudas, insuficiência renal e pulmonar, dependendo da fase de vida. Nas mãos e nos pés principalmente das crianças, pode haver tumefacção causado pela obstrução de vasos naquelas áreas, também acompanhado de dor. Pode ainda ocasionar uma maior suscetibilidade à infecções.
Epidemiologia

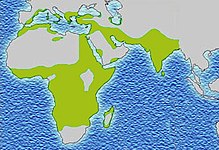
No ocidente, a prevalência é de cerca de 1 em cada 5000, mas em áreas endêmicas da Índia, Arabia Saudita e Nigéria a prevalência varia entre 2 e 22,2%. Nessas regiões é comum encontrar pessoas resistentes a doença, produzindo maior quantidade de hemácias saudáveis.
Em 2001, 90% sobreviviam até os 20 anos e 50% até os 50 anos, sendo muitas das mortes causadas por outras doenças endêmicas da região, como malária e AIDS.
Relação com a malária
Os portadores da anemia falciforme são geralmente mais resistentes à malária do que as pessoas que não têm essa deficiência. Isso ocorre pois os protozoários Plasmodium necessariamente se reproduzem no interior das hemácias humanas e as hemácias danificadas do indivíduo falciforme não são adequadas a esse tipo de função, mesmo quando exposto ao vetor da doença, o mosquito Anopheles contaminado.
Tratamento
A única cura para a anemia falciforme é o transplante de medula óssea. Este tratamento, no entanto, foi realizado em um número relativamente grande de pacientes ao redor do mundo, com maior taxa de sucesso entre crianças. Ainda é necessário um número maior de estudos e a determinação de características clínicas que permitam indicar o transplante com maior segurança. Alguns trabalhos experimentais tem sido feitos com terapia gênica.
Do ponto de vista clínico, o uso de hidroxiuréia, um quimioterápico inibidor da ribonucleotidase vem se revelando útil, por diminuir o número de episódios dolorosos e síndrome torácica aguda. Esta medicação atua por diversos meios, aumentando hemoglobina fetal, diminuindo leucócitos e reticulócitos aderentes ao endotélio e elevando os níveis de óxido nítrico. O uso de hidroxiuréia deve ser feito com supervisão médica, pelo risco de depressão da função da medula óssea e infecções. Além disso os usuários não podem engravidar durante seu uso pelo risco de teratogenicidade para o feto. A experiência clínica de 25 anos com esta medicação não revelou aumento da chance de câncer em seus usuário e trabalhos recentes sugerem aumento da sobrevida dos pacientes.
São realizadas transfusões durante exacerbações da anemia. Pacientes com complicações graves, como acidente vascular cerebral, são submetidos a regimes regulares de transfusão sanguínea ou exsanguineo-transfusão, em geral a cada 28 dias. Pacientes neste regime tendem a acumular ferro no organismo (hemossiderose), o que pode ser controlado com o uso de substâncias quelantes. Se o ferro não for adequadamente quelado pode se depositar em órgãos como fígado e coração trazendo outras complicações.
Durante crises, deve ser administrada hidratação intravenosa e analgesia preferencialmente com opioides. É sugerido que o uso de dolantina, um dos opioides endovenosos, seja evitado, pelo risco maior de dependência.Toda crise dolorosa tem de ser avaliada como prenúncio de complicações graves, como a síndrome torácica aguda. O tratamento deve evitar hiper-hidratação e hiper-sedação e privilegiar a fisioterapia respiratória.
Cura
Anunciado em 12 de junho de 2020, resultado de um estudo preliminar, mostra que o CRISPR fornece 'cura funcional' para pacientes com talassemia beta e doença falciforme. Embora os três pacientes tenham experimentado alguns efeitos adversos devido à quimioterapia, a edição do gene CRISPR parece segura.