
| Gangliosidose | |
|---|---|
 | |
| Especialidade | endocrinologia |
| Classificação e recursos externos | |
| CID-10 | E75.0-E75.1 |
| CID-9 | 330.1 |
| CID-11 | 797306953 |
| MeSH | D005733 |
|
| |
Gangliosidoses são esfingolipidoses (distúrbios de armazenamento lisossômico) de origem genética autossômica recessiva caracterizadas pelo acúmulo generalizado de gangliosídios, oligossacarídeos ou sulfato de queratano mucopolissacarídeos (e seus derivados).
Classificação
Segundo o CID-10 pode ser classificado em:
Causa
Deficiências genéticas autossômicas recessivas afetando igualmente mulheres e homens, podendo os primeiros sintomas aparecem aos primeiros meses de vida (caso severa, tipo 1) ou apenas depois dos 18 meses (caso moderada, tipo 2) ou somente em adultos (caso leve, tipo 3).
- GM1
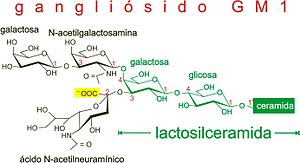
A gangliosidose GM1 é causada por uma deficiência na beta-galactosidase, resultando em acúmulo de materiais lipídicos e ácidos em células dos sistemas nervoso central e periférico, mas em particular nas células nervosas.
- GM2
As gangliosidoses GM2 são um grupo de desordens genéticas relacionadas causadas por uma deficiência da enzima beta-hexosaminidase. Esta enzima catalisa a biodegradação de derivados de ácidos graxos conhecidos como gangliosídios.
Gangliosídios
Glicolípidos são complexos que contêm ácido siálico e que são encontrados especialmente no cérebro. São essenciais para a mielinização, para a integridade dos axônios neuronais e para a transmissão de impulsos nervosos.
Sinais e sintomas
- Gangliosidose GM1 tipo I ou infantil
Geralmente os sintomas só aparecem aos 6 meses de idade, geralmente parecendo normal antes disso, e os primeiros sintomas incluem:
- Desenvolvimento desacelera
- Músculos enfraquecer
- Perder de habilidades
- Sensibilidade a ruídos.
Conforme a doença progride, ocorre:
- Aumento do fígado e do baço (hepatoesplenomegalia)
- Anormalidades esqueléticas
- Convulsões
- Deficiência intelectual
- Cegueira progressiva.
A perda de visão ocorre como o tecido sensível à luz na parte de trás do olho (retina), deteriora-se gradualmente. Um ponto vermelho na retina pode ser identificado com um exame oftalmológico e é característico desta desordem. Em alguns casos, os indivíduos afetados têm características faciais distintivas, gengivas alargada (hipertrofia gengival) e um músculo do coração aumentado e enfraquecido (cardiomiopatia).
- Gangliosidose GM1 tipo 2 ou juvenil
Forma intermediárias da doença, crianças com GM1 tipo II tem desenvolvimento normal até os 18 meses (forma infantil tardia) ou 5 anos (forma juvenil) quando aparecem os seguintes sintomas:
- Regressão do desenvolvimento
- Características faciais distintivas
- Aumento do fígado e do baço
- Gangliosidose GM1 tipo 3 ou adulto
A forma mais branda e rara da doença, a maioria dos indivíduos afetados só desenvolvem sinais e sintomas na adolescência e incluem:
- Enrijecimento involuntário de vários músculos (distonia)
- Anormalidades dos ossos da coluna vertebral (vértebras)
- Características faciais distintas
- Gangliosidose GM2 ou Doença de Tay-Sachs ou Doença de Sandhoff
A criança se desenvolve normalmente até que os sintomas aparecem depois de alguns meses de idade, e os sintomas incluem:
- Declínio mental e de habilidades físicas
- Cegueira
- Surdez
- Incapacidade de engolir
- Atrofia muscular
- Paralisia.
Prevalência
Atingem 1 em cada 100.000 nascidos. O GM1 tipo 1 é mais comum que os outros tipos. O tipo 3 é mais comum entre descendentes de japoneses. É tão comum em homens quanto em mulheres.
Tratamento
Atualmente, nenhum tratamento médico eficaz está disponível para o distúrbio subjacente em pacientes com gangliosidose GM1. Transplante de medula óssea foi bem sucedido em um indivíduo com gangliosidose GM1 infantil e juvenil, no entanto, nenhum benefício a longo prazo foi relatado. O tratamento de algumas sequelas neurológicas está disponível, mas não altera significativamente o curso clínico. Costuma ser fatal alguns meses após o surgimento dos sintomas.
Transplante de células-tronco tem sido defendido como um possível tratamento por causa do sucesso em outras doenças de depósito lisossômico. Pesquisa ativa nas áreas de substituição enzimática e terapia genética para gangliosidose GM1 está em curso, mas não avançou para testes em humanos.
A expectativa de vida no GM1 tipo I é de poucos meses, no tipo II pode chegar a idade adulta e do tipo III pode passar de vinte anos.