
| Doença de Hirschsprung | |
|---|---|
 | |
| Especialidade | pediatria, digestive system surgery, gastroenterologia |
| Classificação e recursos externos | |
| CID-10 | Q43.1 |
| CID-11 | 1772690306 |
| OMIM | 600156, 606874, 606875, 608462, 611644 |
| DiseasesDB | 5901 |
| MedlinePlus | 001140 |
| eMedicine | 178493 |
| MeSH | D006627 |
|
| |
A Doença de Hirschsprung (DH ou HD, na sigla em inglês), megacólon agangliônico congênito ou megacólon congênito é um doença congênita rara a qual ocorre um aumento do cólon causado por obstrução intestinal resultante de uma ausência das células ganglionares do plexo mioentérico e submucoso do sistema nervoso entérico que se inicia no ânus e progride superiormente. O sintoma geralmente se apresentam entre os primeiros dias de vida até dois meses, e o mais proeminente é a constipação, outros sintomas podem incluir vômitos, dor abdominal, diarréia e crescimento lento. As complicações podem incluir enterocolite, megacólon, obstrução intestinal e perfuração intestinal.
O tratamento geralmente é feito por cirurgia para remover a seção afetada do intestino. Em alguns casos, um transplante intestinal pode ser recomendado. A Doença de Hirschsprung ocorre em cerca de um em cada 5 mil recém-nascidos, indivíduos do sexo masculino são afetados com mais frequência. Acredita-se que a condição tenha sido detectada pela primeira vez em 1691 pelo anatomista neerlandês Frederik Ruysch, mas a doença recebe o nome do médico dinamarquês Harald Hirschsprung, que a descreveu em 1888 e iniciou as pesquisas sobre a doença.
Sintomas
Normalmente, a Doença de Hirschsprung é diagnosticada logo após o nascimento devido à presença de megacólon ou porque o bebê não consegue evacuar as primeiras fezes (mecônio). Normalmente, 90% dos bebês passam seu primeiro mecônio em 24 horas e 99% em 48 horas. Outros sintomas incluem vômito verde ou marrom, inchaço do abdômen, gases excessivos e diarreia com sangue.
Alguns casos são diagnosticados mais tarde, na infância, mas geralmente antes dos 10 anos de idade. A criança pode apresentar retenção fecal, constipação ou distensão abdominal.
Apesar de raro, também é possível desenvolver a doença na idade adulta. Também há casos de pessoas que nascem com adoeça mas são diagnosticadas apenas quando adultas.
Síndromes associadas
A Doença de Hirschsprung também pode se apresentar como parte de distúrbios multissistêmicos, como:
- Síndrome de Bardet-Biedl;
- Hipoplasia da Cartilagem Cabelo;
- Síndrome de Hipoventilação Central Congênita;
- Neoplasia Endócrina Múltipla Tipo 2;
- Síndrome de Mowat-Wilson;
- Síndrome de Smith-Lemli-Opitz;
- Trissomia do Cromossomo 21(Síndrome de Down);
- Algumas formas de Síndrome de Waardenburg.
Causa
O distúrbio pode ocorrer por si só ou em associação com outros distúrbios genéticos, como Síndrome de Down ou Síndrome de Waardenburg. Cerca de metade dos casos isolados estão ligados a uma mutação genética específica e cerca de 20% ocorrem dentro da família. Alguns casos ocorrem de maneira autossômica dominante. A causa dos casos restantes não é clara. Se pais saudáveis tiverem um filho com a doença, o próximo filho tem 4% de chance de nascer com a condição. A condição é dividida em dois tipos principais, segmento curto e segmento longo, dependendo de quanto o intestino é afetado. Raramente, o intestino delgado é afetado. O diagnóstico é baseado na avaliação dos sintomas e confirmado por biópsia.
Genética
| Modelo | OMIM | Gene | Locus |
|---|---|---|---|
| HSCR1 | OMIM 142623 | RET | 10q11.2 |
| HSCR2 | OMIM 600155 | EDNRB | 13q22 |
| HSCR3 | OMIM 600837 | GDNF | 5p13.1-p12 |
| HSCR4 | OMIM 131242 | EDN3 | 20q13.2-q13.3 |
| HSCR5 | OMIM 600156 | ? | 21q22 |
| HSCR6 | OMIM 606874 | ? | 3p21 |
| HSCR7 | OMIM 606875 | ? | 19q12 |
| HSCR8 | OMIM 608462 | ? | 16q23 |
| HSCR9 | OMIM 611644 | ? | 4q31-32 |
| - | OMIM 602229 | SOX10 | 22q13 |
| - | OMIM 600423 | ECE1 | 1p36.1 |
| - | OMIM 602018 | NRTN | 19p13.3 |
| - | OMIM 602595 | GEMIN2 (proteína 2 de ligação à gema) | 14q13-q21 |
| - | OMIM 191315 | NTRK1 | 1q23.1 |
| - | OMIM 605802 | ZEB2 | 2q22.3 |
Vários genes e regiões específicas nos cromossomos (Lócus) foram mostrados ou sugeridos como associados à Doença de Hirschsprung:
O proto-oncogene RET é responsável pela maior proporção de casos familiares e esporádicos, com uma ampla gama de mutações espalhadas ao longo de toda a sua região de codificação. Um proto-oncogene pode causar câncer se for mutado ou superexpresso.
Proto-oncogene RET
RET é um gene que codifica proteínas que auxiliam as células da crista neural em seu movimento através do trato digestivo durante o desenvolvimento do embrião. Essas células da crista neural eventualmente formam feixes de células nervosas chamadas gânglios. O EDNRB codifica as proteínas que conectam essas células nervosas ao trato digestivo. Assim, mutações nesses dois genes podem levar diretamente à ausência de certas fibras nervosas no cólon. A pesquisa sugere que vários genes estão associados à Doença de Hirschsprung. Além disso, uma nova pesquisa sugere que as mutações nas sequências genômicas envolvidas na regulação do EDNRB têm um impacto maior na Doença de Hirschsprung do que se pensava anteriormente.
O gene RET pode sofrer mutações de várias maneiras e está associado à Síndrome de Down. Visto que a Síndrome de Down é comórbida em 2% dos casos de Hirschsprung, existe uma probabilidade de que o RET esteja fortemente envolvido na Doença de Hirschsprung e na Síndrome de Down. O RET também está associado ao câncer medular da tireoide e ao neuroblastoma, um tipo de câncer comum em crianças. Ambos os distúrbios são mais comuns em pacientes de Hirschsprung do que na população em geral. Uma função que RE T controla é a viagem das células da crista neural através dos intestinos no feto em desenvolvimento. Quanto mais cedo a mutação RET ocorre na Doença de Hirschsprung, mais grave se torna a doença.
Outros genes
Variações de DNA comuns e raras na Neuregulina 1(NRG1) e na NRG3 (NRG3) foram inicialmente mostradas como associadas à doença em pacientes chineses por meio de um estudo de associação do genoma realizado pela equipe de Hong Kong em 2009 e 2012, respectivamente. Estudos subsequentes em pacientes asiáticos e caucasianos confirmaram os achados iniciais da Universidade de Hong Kong. Variantes raras e comuns nesses dois genes foram identificadas em tailandeses, coreanos, indonésios e espanhóis. Esses dois genes são conhecidos por desempenhar um papel na formação do sistema nervoso entérico; portanto, é provável que estejam envolvidos na patologia da Doença de Hirschsprung, pelo menos em alguns casos.
Outro gene associado a esta condição é a NADPH oxidase, domínio 5 de ligação de cálcio de mão EF (NOX5). Este gene está localizado no braço longo do cromossomo 15 (15q23).
Fisiopatologia
Durante o desenvolvimento pré-natal normal, as células da crista neural migram para o intestino grosso (cólon) para formar as redes de nervos chamadas plexo mioentérico (Plexo de Auerbach) (entre as camadas de músculo liso da parede do trato gastrointestinal) e o plexo submucoso (Plexo de Meissner) (dentro da submucosa da parede do trato gastrointestinal). Na Doença de Hirschsprung, a migração não é completa e parte do cólon não possui esses corpos nervosos que regulam a atividade do cólon. O segmento afetado do cólon não consegue relaxar e passar fezes, criando uma obstrução.
A teoria mais aceita é que a Doença de Hirschsprung seja causada por um defeito na migração craniocaudal de neuroblastos originados da crista neural que ocorre durante as primeiras 12 semanas de gestação. Defeitos na diferenciação de neuroblastos em células ganglionares e destruição acelerada de células ganglionares dentro do intestino também podem contribuir para o distúrbio.
A falta de células ganglionares no plexo mioentérico e submucoso está associada à Doença de Hirschsprung. Com a Doença de Hirschsprung, o segmento sem neurônios (aganglionar) fica contraído, fazendo com que a seção proximal normal do intestino se distenda com fezes. Acredita-se que esse estreitamento do cólon distal e a falha de relaxamento no segmento aganglionar sejam causados pela falta de neurônios contendo óxido nítrico sintase.
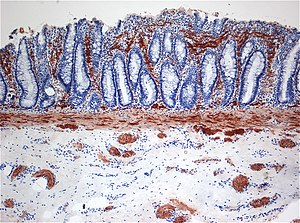
A característica mais citada é a ausência de células ganglionares: notadamente em homens, 75% não têm nenhuma no final do cólon (retossigmóide) e 8% não têm células ganglionares em todo o cólon. A seção aumentada do intestino é encontrada proximalmente, enquanto a seção aganglionar estreitada é encontrada distalmente, mais perto do final do intestino. A ausência de células ganglionares resulta em uma superestimulação persistente dos nervos na região afetada, resultando em contração.
A doença equivalente em cavalos é a Síndrome Letal Do Overo Branco.
Diagnóstico

O diagnóstico definitivo é feito por biópsia por sucção do segmento estreitado distalmente. Um exame histológico do tecido tem por objetivo mostrar a falta de células nervosas ganglionares. As técnicas de diagnóstico envolvem manometria anorretal,enema baritado e biópsia retal. A biópsia retal por sucção é considerada o padrão ouro internacional atual no diagnóstico da Doença de Hirschsprung.
Exames radiológicos também podem ajudar no diagnóstico. A cineanografia (fluoroscopia do meio de contraste passando pela região anorretal) auxilia na determinação do nível dos intestinos afetados.
Tratamento
O Tratamento da doença de Hirschsprung consiste na remoção cirúrgica (ressecção) da seção anormal do cólon, seguida de anastomose .
Colostomia
O primeiro estágio do tratamento costumava ser uma colostomia reversível. Nessa abordagem, a extremidade saudável do intestino grosso é cortada e presa a uma abertura criada na parte frontal do abdômen. As fezes serão descartadas através de um orifício no abdômen, chamada colostomia, para uma bolsa de ostomia. Mais tarde, após a recuperação do paciente e do intestino, quando o peso e a idade estiverem ideais, a "nova" extremidade funcional do intestino é conectada ao ânus. O primeiro tratamento cirúrgico envolvendo ressecção cirúrgica seguida de reanastomose sem colostomia ocorreu já em 1933 pelo Dr. Baird em Birmingham em um menino de um ano de idade.
Ilestomia

Em casos raros, quando uma grande parte, ou todo o intestino grosso é afetado, quando outras doenças se associam e impedem a recuperação e o tratamento esperado com colostomia, é necessária uma ileostomia. O ileostomia irá ignorar todo o intestino grosso e as fezes serão eliminadas diretamente do intestino delgado para a bolsa de ileostomia.
Outros procedimentos
O cirurgião sueco-estadunidense Orvar Swenson (1909–2012), que descobriu a causa da Doença de Hirschsprung, realizou pela primeira vez o tratamento cirúrgico da doença atraves de cirurgia pull-through, em 1948. Desde então, o procedimento tem sido usado, principalmente, em pacientes mais jovens. O procedimento consistem em puxar o intestino para fora do ânus, cortar a parte afetada pela doença e conectar a parte saudável no ânus.

Atualmente, várias abordagens cirúrgicas diferentes são usadas, incluindo os procedimentos de Swenson, Soave, Duhamel e Boley. O procedimento de Swenson deixa uma pequena porção do intestino doente. O procedimento Soave, em homenagem ao cirurgião pediatra italiano, Franco Soave [it] (1917–1984), deixa a parede externa do cólon inalterada. O procedimento de Boley, iniciado pelo cirurgião estadunidense Scott Boley (n. 1941), é uma pequena modificação do procedimento de Soave, então o procedimento é constantemente chamado de "Soave-Boley". O procedimento de Duhamel, em homenagem ao cirurgião pediátrico francês Bernard Duhamel (1917–1996), usa um grampeador cirúrgico para conectar o intestino saudável ao doente.
15% das crianças não conseguem o controle intestinal completo, então outros tratamentos são necessários. A constipação pode ser tratada com laxantes ou dieta rica em fibras. Nesses pacientes, a desidratação grave pode ser um fator importante em seu estilo de vida. A ileostomia e o Enema Colônico Anterógrado de Malone também é uma opção. A Técnica de Malone consiste em passar um tubo pela parede abdominal para o apêndice ou, se disponível, para o cólon. O intestino é então irrigado diariamente.
Se a porção afetada do intestino grosso estiver restrita à porção inferior do reto, outros procedimentos cirúrgicos podem ser realizados, como uma miectomia retal posterior. O prognóstico tem resultados positivos em 70% dos casos. A constipação crônica pós-operatória está presente em 7 a 8% dos casos operados. Enterocolite pós-operatória, uma manifestação grave, está presente em 10–20% dos pacientes operados.
Epidemiologia
De acordo com um estudo de 1984 conduzido em Maryland, a Doença de Hirschsprung aparece em 18,6 por 100.000 nascidos vivos. No Japão, ocorre a uma taxa semelhante de cerca de 1 em 5.000 nascimentos (20 por 100.000). A doença também mais comum em indivíduos do sexo masculino e em pessoas brancas. 9% casos de Hirschsprung ocorrem em Pessoas com Síndrome de Down. A maioria dos casos é diagnosticada antes de o paciente completar 10 anos de idade.
História
O primeiro relato da doença de Hirschsprung data de 1691, quando foi descrita pelo anatomista neerlandês Frederik Ruysch. No entanto, a doença foi batizada em homenagem a Harald Hirschsprung, o médico dinamarquês que descreveu pela primeira vez dois bebês que morreram dessa doença em 1888.
A Doença de Hirschsprung é uma doença congênita do cólon em que certas células nervosas, conhecidas como células ganglionares, estão ausentes, causando constipação crônica. Em pacientes com Doença de Hirschsprung, os plexos mioentérico e submucoso estão ausentes.
A primeira publicação sobre uma importante descoberta genética da doença foi de Martucciello Giuseppe em 1992. Os autores descreveram o caso de um paciente com aganglionose colônica total associada a um cariótipo 46, XX, del 10 (q11.21 q21.2). O principal gene da Doença de Hirschsprung foi identificado nesta região cromossômica 10, era o proto-oncogene RET.
Por muito tempo, a Doença de Hirschsprung foi considerada uma doença multifatorial, em que uma combinação de fatores naturais e estímulos era considerada a causa. No entanto, em agosto de 1993, dois artigos de grupos independentes na Nature Genetics apontou que a Doença de Hirschsprung poderia ser mapeada para um trecho do cromossomo 10. Essa pesquisa também sugeriu que um único gene era responsável pelo distúrbio. No entanto, os pesquisadores não conseguiram isolá-lo.
Veja também
- Acalasia
- Ostomia
- Ileostomia
- Colostomia
- Íleo paralítico
- Elise Sørensen
- Bolsa de ostomia
- Harald Hirschsprung
- Displasia neuronal intestinal